Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
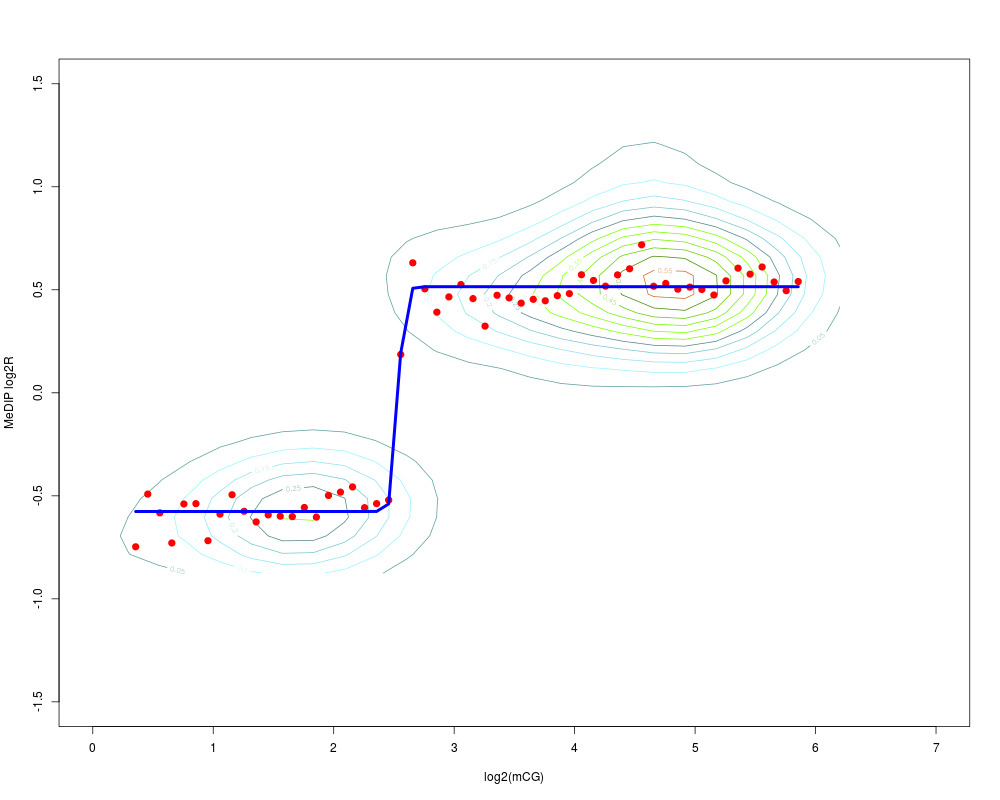
Determining the logistic model of MeDIP enrichment in respect to the expected DNA methylation levelDescriptionProbe-level MeDIP weighted enrichment is compared to the expected DNA methytlation level. The former is determined applying MeDIP protocol to a fully methylated DNA. The latter is determined as the count of CpGs for each probe. This is assumed to be the methylation level of each probe in a fully methylated sample. UsageMEDME(data, sample, CGcountThr = 1, figName = NULL) Arguments
DetailsThe model should be applied on calibration data containing MeDIP enrichment of fully methylated DNA, most likely artificially generated (see references). Nevertheless, in case chromosome or genome-wide human tiling arrays are used a regular sample could be used too. In fact, human genomic DNA is known to be hyper-methylated but in the promoter regions. Of course the performance of the method is expected to be somehow affected by this approximation. ValueThe logistic model as returned from the multdrc function from the drc R library Referenceshttp://genome.cshlp.org/cgi/content/abstract/gr.080721.108v1 See Also
Examplesdata(testMEDMEset) ## just an example with the first 1000 probes testMEDMEset = smooth(data = testMEDMEset[1:1000, ]) library(BSgenome.Hsapiens.UCSC.hg18) testMEDMEset = CGcount(data = testMEDMEset) MEDMEmodel = MEDME(data = testMEDMEset, sample = 1, CGcountThr = 1, figName = NULL) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MEDME)
Attaching package: 'MEDME'
The following object is masked from 'package:stats':
smooth
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/MEDME/MEDME.Rd_%03d_medium.png", width=480, height=480)
> ### Name: MEDME
> ### Title: Determining the logistic model of MeDIP enrichment in respect to
> ### the expected DNA methylation level
> ### Aliases: MEDME
>
> ### ** Examples
>
> data(testMEDMEset)
> ## just an example with the first 1000 probes
> testMEDMEset = smooth(data = testMEDMEset[1:1000, ])
chrX
> library(BSgenome.Hsapiens.UCSC.hg18)
Loading required package: BSgenome
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Attaching package: 'GenomicRanges'
The following object is masked from 'package:MEDME':
pos
Loading required package: Biostrings
Loading required package: XVector
Loading required package: rtracklayer
> testMEDMEset = CGcount(data = testMEDMEset)
chrX
> MEDMEmodel = MEDME(data = testMEDMEset, sample = 1, CGcountThr = 1, figName = NULL)
>
>
>
>
>
> dev.off()
null device
1
>
|