Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
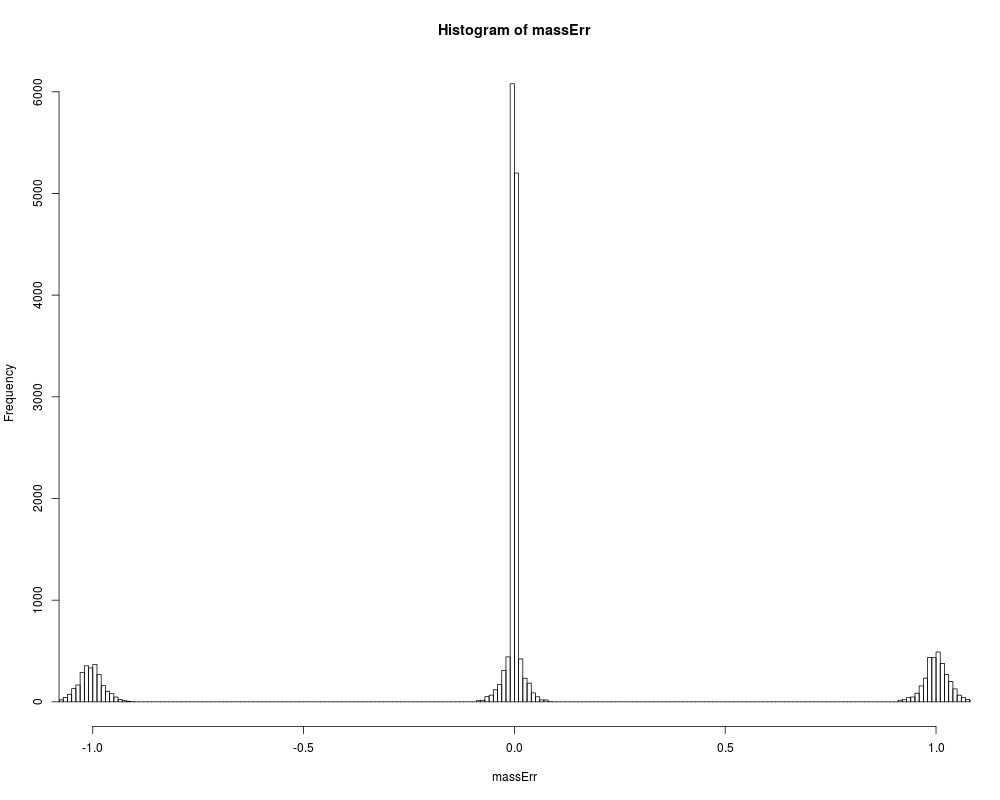
Corrects wrong selection of monoisotopic peakDescriptionIn a typical setting instruments select ions for fragmentation primarily based on ion intensity. For low molecular weight peptides the most intense peak usually corresponds to monoisotopic peak (that is only C12 carbon isotopes). With increase of molecular weight, instensity of monoisotopic peak becomes smaller relatively to heavier peptide isotopes (that is containing one or a few C13 isotopes). The function subtracts or adds the mass difference between C13 and C12 isotopes (1.0033548378 Da) if that reduces the mass error. Such a mass error arises from the fact that instrument may peak non-monoisotopic peak for fragmentation and thus report the mass that is different by ~ 1 Da. Usage
correct_peak_selection(object)
Arguments
ValueReturns an instance of "MSnID" class with updated
Author(s)Vladislav A Petyuk vladislav.petyuk@pnnl.gov See Also
Examples
data(c_elegans)
# plot original mass error
massErr <- (msnidObj$experimentalMassToCharge -
msnidObj$calculatedMassToCharge) *
msnidObj$chargeState
hist(massErr,xlim=c(-1,+1), breaks=seq(-1.5,+1.5,0.01))
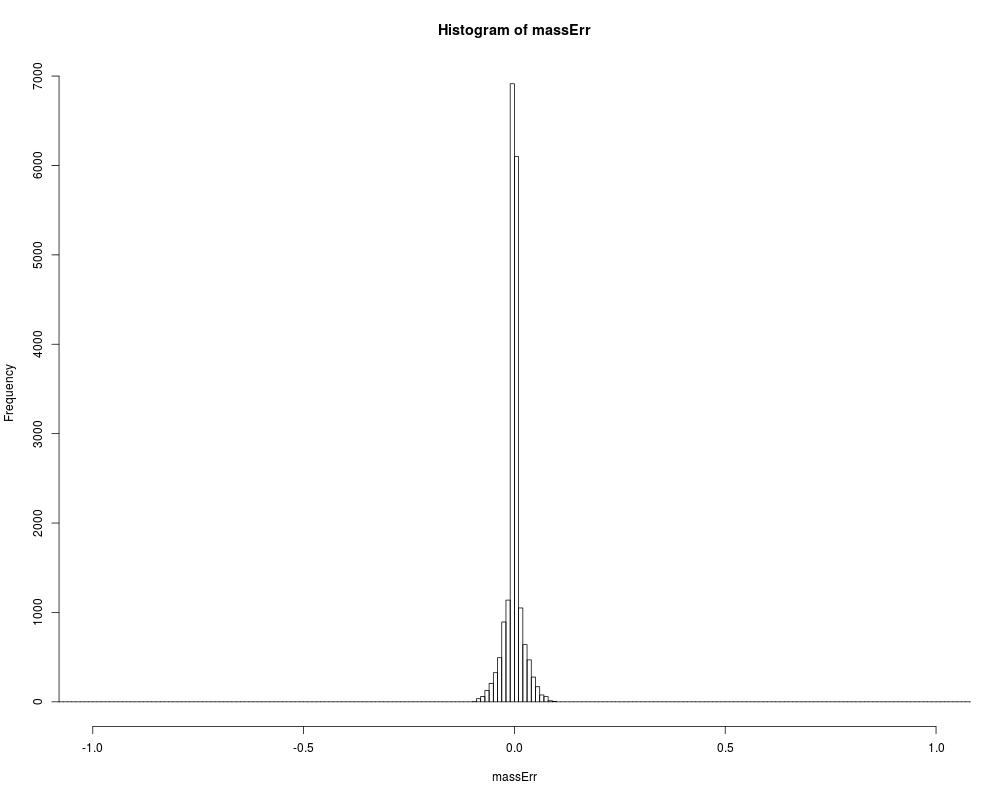
# fixing the problem of picking wrong monoisotopic peak
msnidObj <- correct_peak_selection(msnidObj)
# plot fixed mass error
massErr <- (msnidObj$experimentalMassToCharge -
msnidObj$calculatedMassToCharge) *
msnidObj$chargeState
hist(massErr,xlim=c(-1,+1), breaks=seq(-1.5,+1.5,0.01))
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MSnID)
Loading required package: Rcpp
Warning messages:
1: replacing previous import 'reshape2::melt' by 'data.table::melt' when loading 'MSnID'
2: replacing previous import 'reshape2::dcast' by 'data.table::dcast' when loading 'MSnID'
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/MSnID/correct_peak_selection.Rd_%03d_medium.png", width=480, height=480)
> ### Name: correct_peak_selection
> ### Title: Corrects wrong selection of monoisotopic peak
> ### Aliases: correct_peak_selection
>
> ### ** Examples
>
> data(c_elegans)
> # plot original mass error
> massErr <- (msnidObj$experimentalMassToCharge -
+ msnidObj$calculatedMassToCharge) *
+ msnidObj$chargeState
> hist(massErr,xlim=c(-1,+1), breaks=seq(-1.5,+1.5,0.01))
> # fixing the problem of picking wrong monoisotopic peak
> msnidObj <- correct_peak_selection(msnidObj)
> # plot fixed mass error
> massErr <- (msnidObj$experimentalMassToCharge -
+ msnidObj$calculatedMassToCharge) *
+ msnidObj$chargeState
> hist(massErr,xlim=c(-1,+1), breaks=seq(-1.5,+1.5,0.01))
>
>
>
>
>
> dev.off()
null device
1
>
|