Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Probability of sharing of rare variants in a family sample within a geneDescriptionComputing probability of sharing of rare variants in a family sample within a genomic region such as a gene. UsageRVgene(ped.mat,ped.listfams,sites,fams,pattern.prob.list,nequiv.list,N.list, type="alleles",minor.allele.vec,precomputed.prob=list(0)) Arguments
Details The function extracts the carriers of the minor allele at each entry in ValueA list with items:
Author(s)Alexandre Bureau <alexandre.bureau@msp.ulaval.ca> References[1] http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#ped [2] Bureau, A., Younkin, S., Parker, M.M., Bailey-Wilson, J.E., Marazita, M.L., Murray, J.C., Mangold, E., Albacha-Hejazi, H., Beaty, T.H. and Ruczinski, I. (2014) Inferring rare disease risk variants based on exact probabilities of sharing by multiple affected relatives. Bioinformatics, 30(15): 2189-96, doi:10.1093/bioinformatics/btu198. See Also
Examples
data(ped.list)
data(ex.ped.mat)
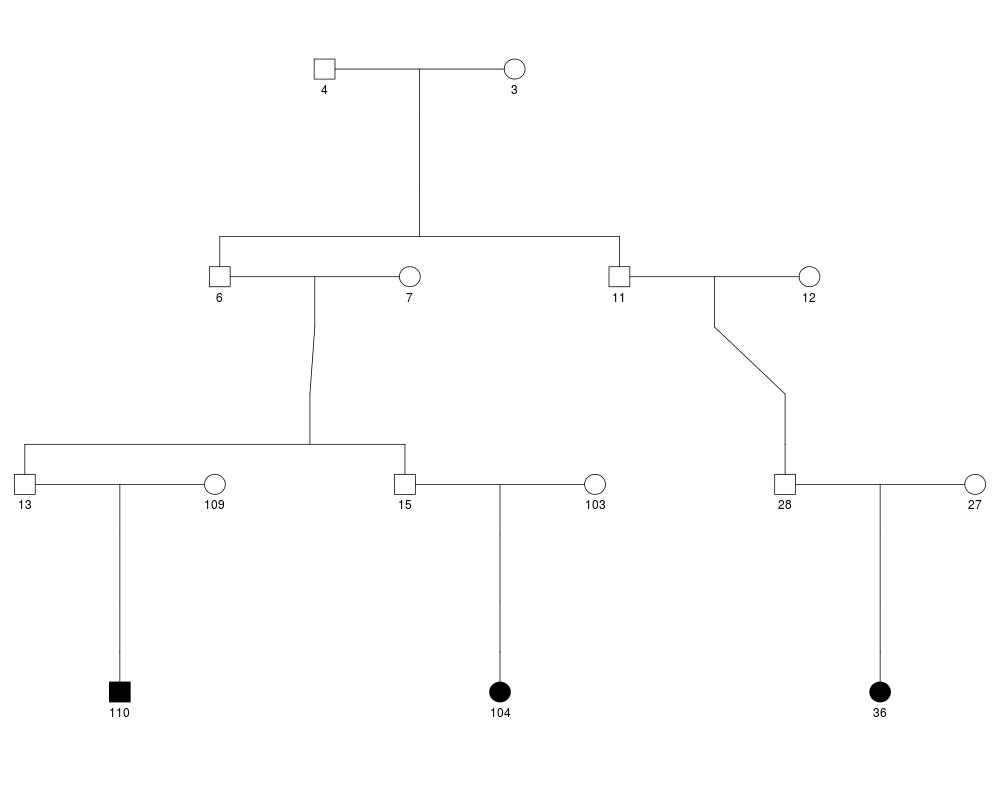
plot(ped.list[[49]])
# Computation of RV sharing probability for 5 sharing patterns in family 28003
fam28003.pattern.prob = c(RVsharing(ped.list[[49]],carriers=c("36","104","110"))@pshare,
RVsharing(ped.list[[49]],carriers=c("36","104"))@pshare,
RVsharing(ped.list[[49]],carriers=c("104","110"))@pshare,
RVsharing(ped.list[[49]],carriers=c("36"))@pshare,
RVsharing(ped.list[[49]],carriers=c("104"))@pshare)
fam28003.nequiv = c(1,2,1,1,2)
# check that distribution sums to 1
sum(fam28003.pattern.prob*fam28003.nequiv)
fam28003.N = c(3,2,2,1,1)
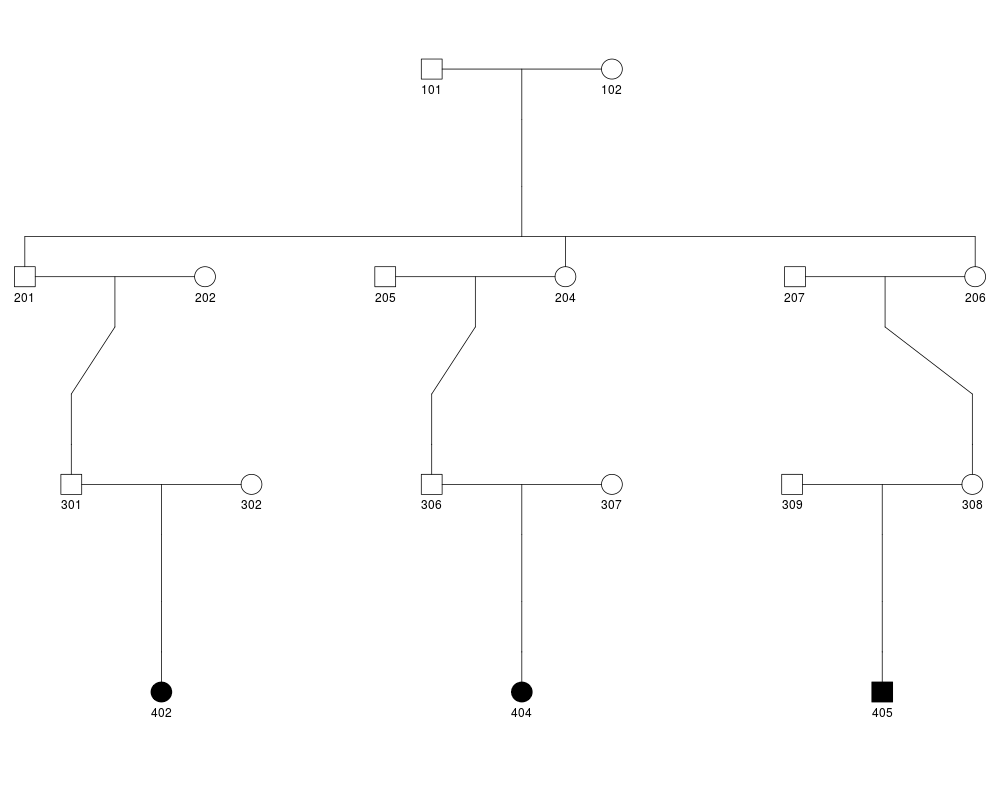
plot(ped.list[[13]])
# Computation of RV sharing probability for 3 sharing patterns in family 15157
fam15157.pattern.prob = c(RVsharing(ped.list[[13]],carriers=c("402","404","405"))@pshare,
RVsharing(ped.list[[13]],carriers=c("402","404"))@pshare,
RVsharing(ped.list[[13]],carriers=c("402"))@pshare)
fam15157.nequiv = c(1,3,3)
# check that distribution sums to 1
sum(fam15157.pattern.prob*fam15157.nequiv)
fam15157.N = 3:1
# Creating lists
ex.pattern.prob.list = list("15157"=fam15157.pattern.prob,"28003"=fam28003.pattern.prob)
ex.nequiv.list = list("15157"=fam15157.nequiv,"28003"=fam28003.nequiv)
ex.N.list = list("15157"=fam15157.N,"28003"=fam28003.N)
ex.ped.obj = ped.list[c(13,49)]
names(ex.ped.obj) = c("15157","28003")
sites = c(92,119)
minor.allele.vec=c(1,4)
RVgene(ex.ped.mat,ex.ped.obj,sites,pattern.prob.list=ex.pattern.prob.list,
nequiv.list=ex.nequiv.list,N.list=ex.N.list,minor.allele.vec=minor.allele.vec)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(RVsharing)
Welcome to RVsharing version 1.6.0
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/RVsharing/RVgene.Rd_%03d_medium.png", width=480, height=480)
> ### Name: RVgene
> ### Title: Probability of sharing of rare variants in a family sample
> ### within a gene
> ### Aliases: RVgene
>
> ### ** Examples
>
> data(ped.list)
> data(ex.ped.mat)
> plot(ped.list[[49]])
> # Computation of RV sharing probability for 5 sharing patterns in family 28003
> fam28003.pattern.prob = c(RVsharing(ped.list[[49]],carriers=c("36","104","110"))@pshare,
+ RVsharing(ped.list[[49]],carriers=c("36","104"))@pshare,
+ RVsharing(ped.list[[49]],carriers=c("104","110"))@pshare,
+ RVsharing(ped.list[[49]],carriers=c("36"))@pshare,
+ RVsharing(ped.list[[49]],carriers=c("104"))@pshare)
> fam28003.nequiv = c(1,2,1,1,2)
> # check that distribution sums to 1
> sum(fam28003.pattern.prob*fam28003.nequiv)
[1] 1
> fam28003.N = c(3,2,2,1,1)
>
> plot(ped.list[[13]])
> # Computation of RV sharing probability for 3 sharing patterns in family 15157
> fam15157.pattern.prob = c(RVsharing(ped.list[[13]],carriers=c("402","404","405"))@pshare,
+ RVsharing(ped.list[[13]],carriers=c("402","404"))@pshare,
+ RVsharing(ped.list[[13]],carriers=c("402"))@pshare)
> fam15157.nequiv = c(1,3,3)
> # check that distribution sums to 1
> sum(fam15157.pattern.prob*fam15157.nequiv)
[1] 1
> fam15157.N = 3:1
>
> # Creating lists
> ex.pattern.prob.list = list("15157"=fam15157.pattern.prob,"28003"=fam28003.pattern.prob)
> ex.nequiv.list = list("15157"=fam15157.nequiv,"28003"=fam28003.nequiv)
> ex.N.list = list("15157"=fam15157.N,"28003"=fam28003.N)
> ex.ped.obj = ped.list[c(13,49)]
> names(ex.ped.obj) = c("15157","28003")
>
> sites = c(92,119)
> minor.allele.vec=c(1,4)
>
> RVgene(ex.ped.mat,ex.ped.obj,sites,pattern.prob.list=ex.pattern.prob.list,
+ nequiv.list=ex.nequiv.list,N.list=ex.N.list,minor.allele.vec=minor.allele.vec)
$p
[1] 0.000159884
$pall
[1] 0.001342282
>
>
>
>
>
> dev.off()
null device
1
>
|