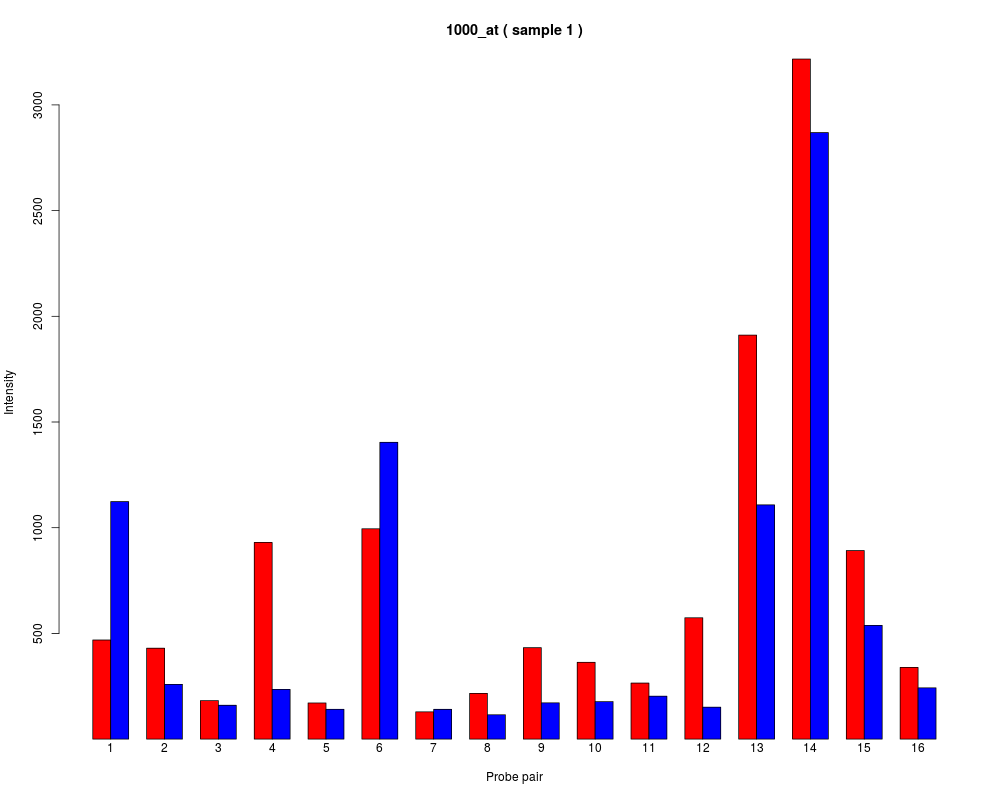
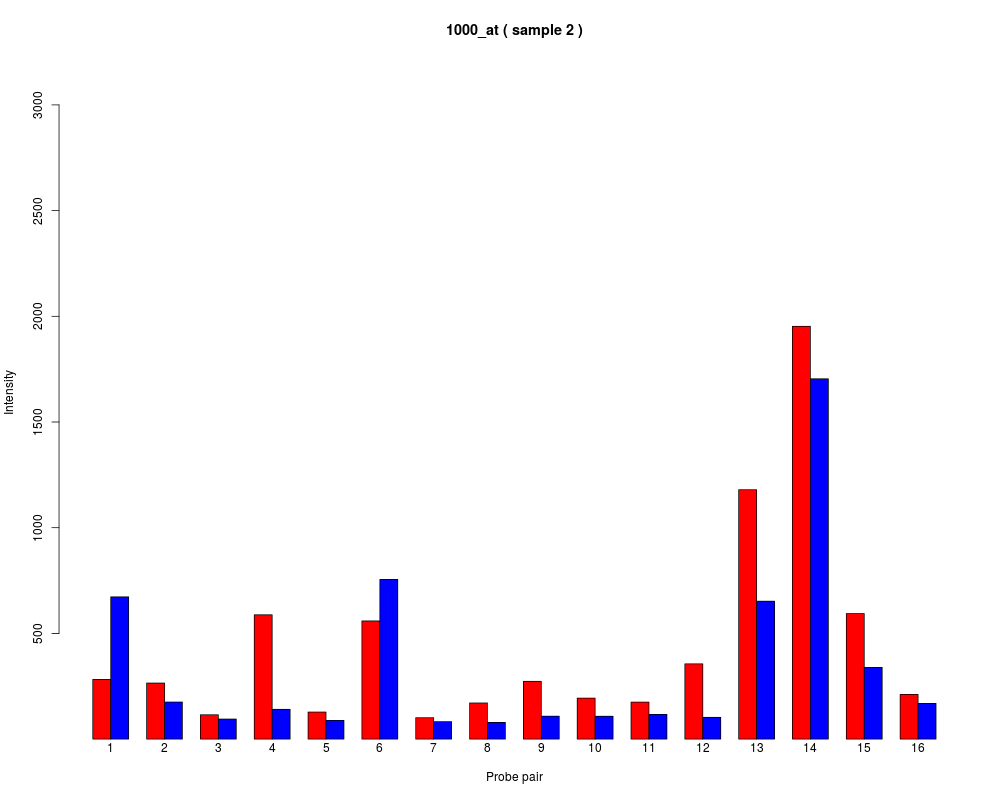
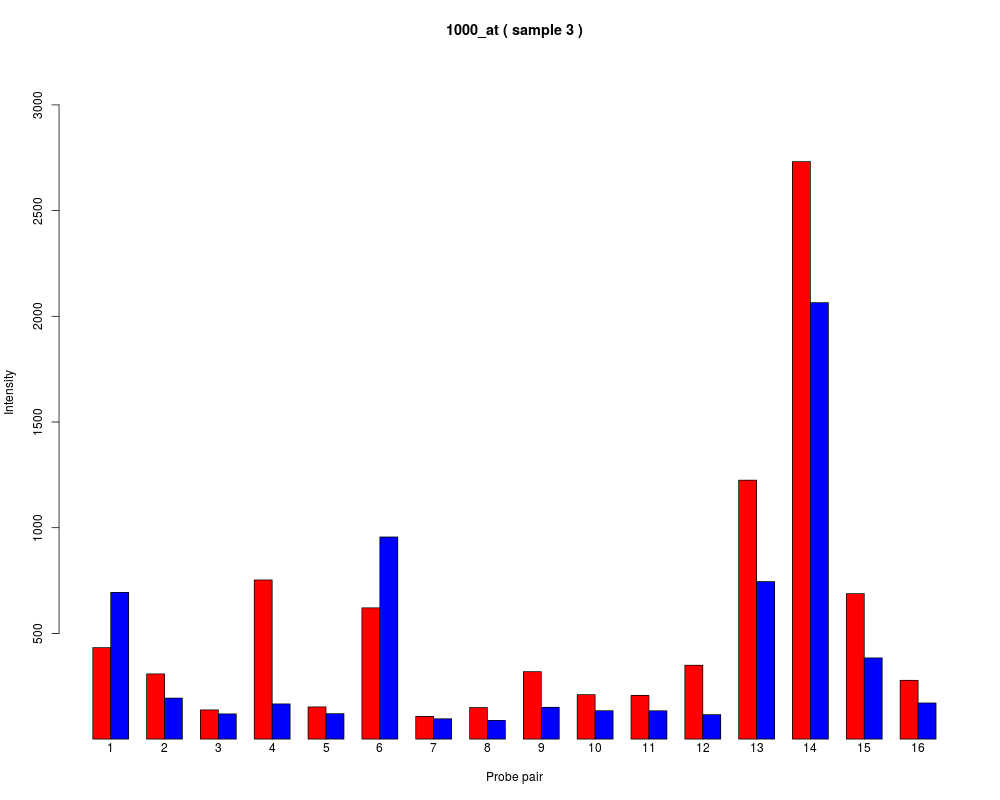
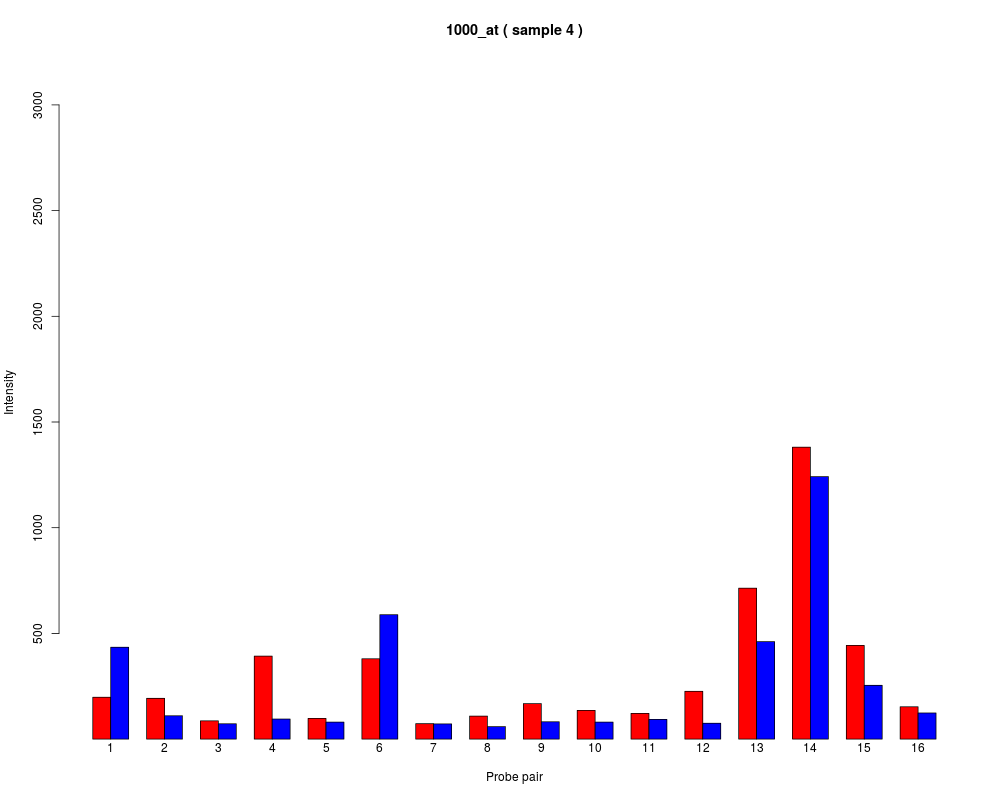
names to be plotted below each bar or group of bars.
ask
ask before ploting the next barplot.
scale
put all the barplot to the same scale.
...
extra parameters to be passed to barplot.
Examples
if (require(affydata)) {
data(Dilution)
gn <- geneNames(Dilution)
pps <- probeset(Dilution, gn[1])[[1]]
barplot.ProbeSet(pps)
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(affy)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/affy/barplot.ProbeSet.Rd_%03d_medium.png", width=480, height=480)
> ### Name: barplot.ProbeSet
> ### Title: show a ProbeSet as barplots
> ### Aliases: barplot.ProbeSet
> ### Keywords: hplot
>
> ### ** Examples
>
> if (require(affydata)) {
+ data(Dilution)
+ gn <- geneNames(Dilution)
+ pps <- probeset(Dilution, gn[1])[[1]]
+
+ barplot.ProbeSet(pps)
+ }
Loading required package: affydata
Package LibPath Item
[1,] "affydata" "/home/ddbj/local/lib64/R/library" "Dilution"
Title
[1,] "AffyBatch instance Dilution"
Warning messages:
1: replacing previous import 'AnnotationDbi::tail' by 'utils::tail' when loading 'hgu95av2cdf'
2: replacing previous import 'AnnotationDbi::head' by 'utils::head' when loading 'hgu95av2cdf'
>
>
>
>
>
> dev.off()
null device
1
>
 .
.