Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
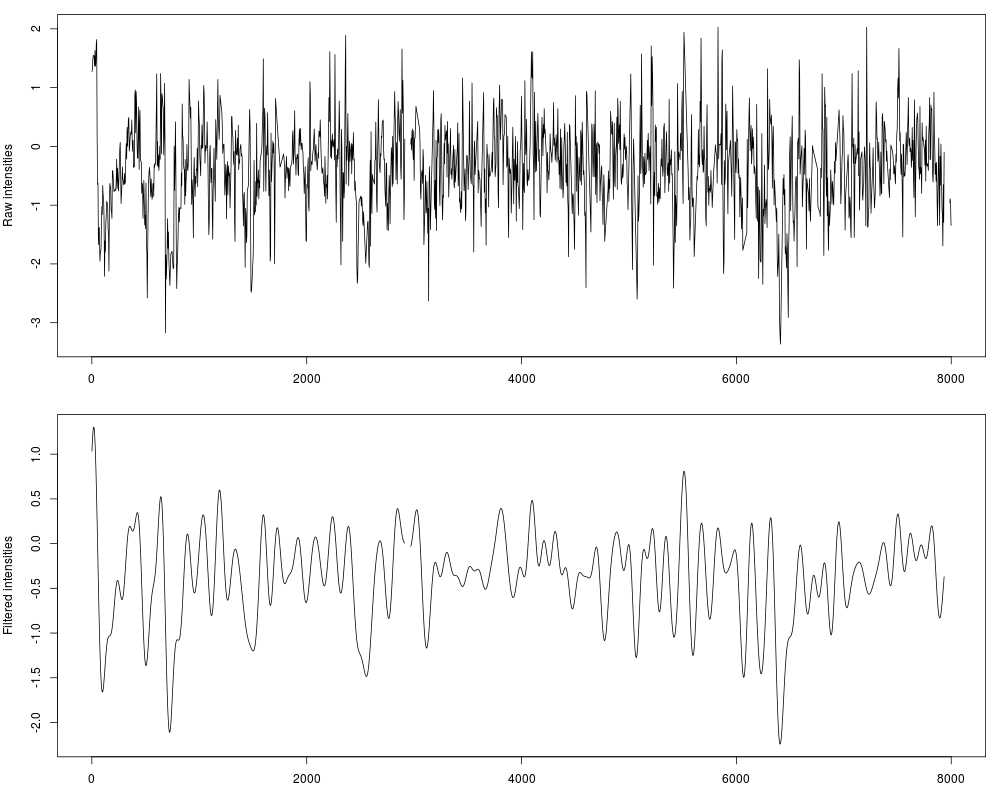
Clean noise and smoothing for genomic data using Fourier-analysisDescriptionRemove noise from genomic data smoothing and cleaning the observed signal. This function doesn't alter the shape or the values of the signal as much as the traditional method of sliding window average does, providing a great correlation within the original and filtered data (>0.99). Usage## S4 method for signature 'SimpleRleList' filterFFT(data, pcKeepComp="auto", showPowerSpec=FALSE, useOptim=TRUE, mc.cores=1, ...) ## S4 method for signature 'list' filterFFT(data, pcKeepComp="auto", showPowerSpec=FALSE, useOptim=TRUE, mc.cores=1, ...) ## S4 method for signature 'Rle' filterFFT(data, pcKeepComp="auto", showPowerSpec=FALSE, useOptim=TRUE, ...) ## S4 method for signature 'numeric' filterFFT(data, pcKeepComp="auto", showPowerSpec=FALSE, useOptim=TRUE, ...) Arguments
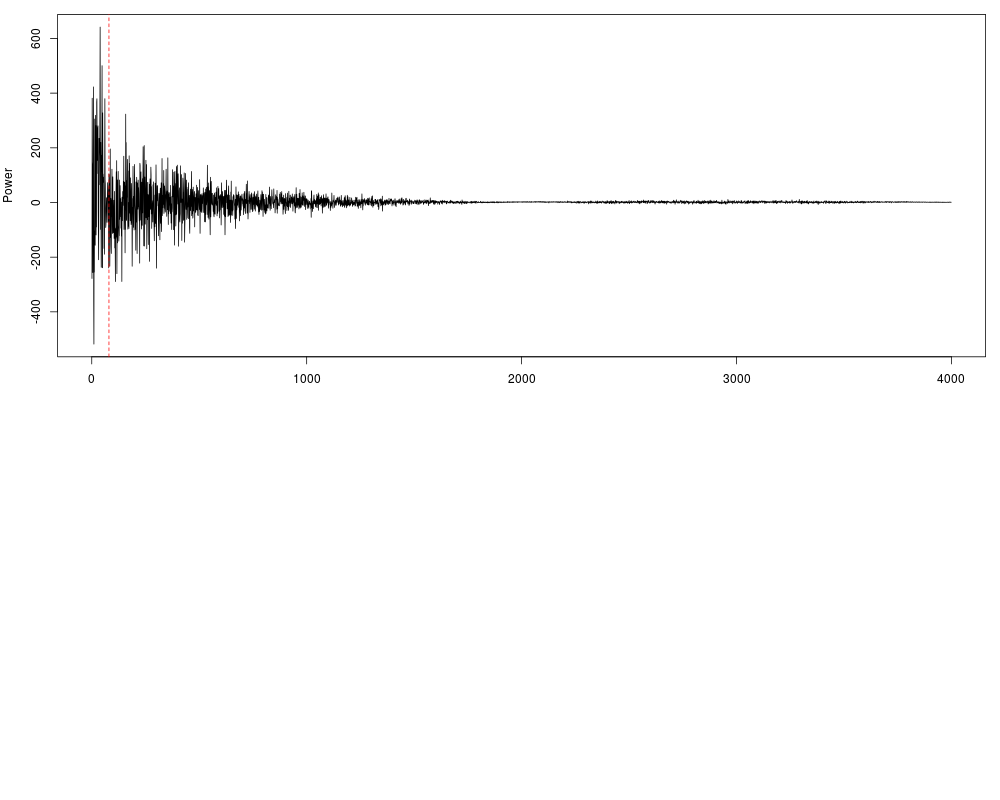
DetailsFourier-analysis principal components selection is widely used in signal processing theory for an unbiased cleaning of a signal over the time. Other procedures, as the traditional sliding window average, can change too much the shape of the results in function of the size of the window, and moreover they don't only smooth the noise without removing it. With a Fourier Transform of the original signal, the input signal is descomposed in diferent wavelets and described as a combination of them. Long frequencies can be explained as a function of two ore more periodical shorter frequecies. This is the reason why long, unperiodic sequences are usually identified as noise, and therefore is desireable to remove them from the signal we have to process. This procedure here is applied to genomic data, providing a novel method to obtain perfectly clean values wich allow an efficient detection of the peaks which can be used for a direct nucleosome position recognition. This function select a certain number of components in the original power spectrum (the result of the Fast Fourier Transform which can be seen with The amout of components to keep (given as a percentage of the input lenght) can be set by the This library also allows the automatic detection of a fitted value for One of the most powerful features of If for some reason you want to apply the function without any kind of optimizations you can specify the parameter ValueNumeric vector with cleaned/smoothed values Author(s)Oscar Flores oflores@mmb.pcb.ub.es, David Rosell david.rossell@irbbarcelona.org ReferencesSmith, Steven W. (1999), The Scientist and Engineer's Guide to Digital Signal Processing (Second ed.), San Diego, Calif.: California Technical Publishing, ISBN 0-9660176-3-3 (availabe online: http://www.dspguide.com/pdfbook.htm) Examples#Load example data, raw hybridization values for Tiling Array raw_data = get(data(nucleosome_tiling)) #Filter data fft_data = filterFFT(raw_data, pcKeepComp=0.01) #See both profiles par(mfrow=c(2,1), mar=c(3, 4, 1, 1)) plot(raw_data, type="l", xlab="position", ylab="Raw intensities") plot(fft_data, type="l", xlab="position", ylab="Filtered intensities") #The power spectrum shows a visual representation of the components fft_data = filterFFT(raw_data, pcKeepComp=0.01, showPowerSpec=TRUE) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(nucleR)
Loading required package: ShortRead
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: BiocParallel
Loading required package: Biostrings
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: GenomicAlignments
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/nucleR/filterFFT.Rd_%03d_medium.png", width=480, height=480)
> ### Name: filterFFT
> ### Title: Clean noise and smoothing for genomic data using
> ### Fourier-analysis
> ### Aliases: filterFFT filterFFT,SimpleRleList-method filterFFT,Rle-method
> ### filterFFT,list-method filterFFT,numeric-method
> ### Keywords: manip
>
> ### ** Examples
>
> #Load example data, raw hybridization values for Tiling Array
> raw_data = get(data(nucleosome_tiling))
>
> #Filter data
> fft_data = filterFFT(raw_data, pcKeepComp=0.01)
>
> #See both profiles
> par(mfrow=c(2,1), mar=c(3, 4, 1, 1))
> plot(raw_data, type="l", xlab="position", ylab="Raw intensities")
> plot(fft_data, type="l", xlab="position", ylab="Filtered intensities")
>
> #The power spectrum shows a visual representation of the components
> fft_data = filterFFT(raw_data, pcKeepComp=0.01, showPowerSpec=TRUE)
>
>
>
>
>
> dev.off()
null device
1
>
|