Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Nucleosome positioning package for RDescriptionNucleosome positioning from Tiling Arrays and High-Troughput Sequencing Experiments Details
This package provides a convenient pipeline to process and analize nucleosome positioning experiments from High-Troughtput Sequencing or Tiling Arrays. Despite it's use is intended to nucleosome experiments, it can be also useful for general ChIP experiments, such as ChIP-on-ChIP or ChIP-Seq. See following example for a brief introduction to the available functions Author(s)Oscar Flores Ricard Illa Maintainer: Ricard Illa <ricard.illa@irbbarcelona.org> Examples
#Load example dataset:
# some NGS paired-end reads, mapped with Bowtie and processed with R
# it is a RangedData object with the start/end coordinates for each read.
reads = get(data(nucleosome_htseq))
#Process the paired end reads, but discard those with length > 200
preads_orig = processReads(reads, type="paired", fragmentLen=200)
#Process the reads, but now trim each read to 40bp around the dyad
preads_trim = processReads(reads, type="paired", fragmentLen=200, trim=40)
#Calculate the coverage, directly in reads per million (r.p.m)
cover_orig = coverage.rpm(preads_orig)
cover_trim = coverage.rpm(preads_trim)
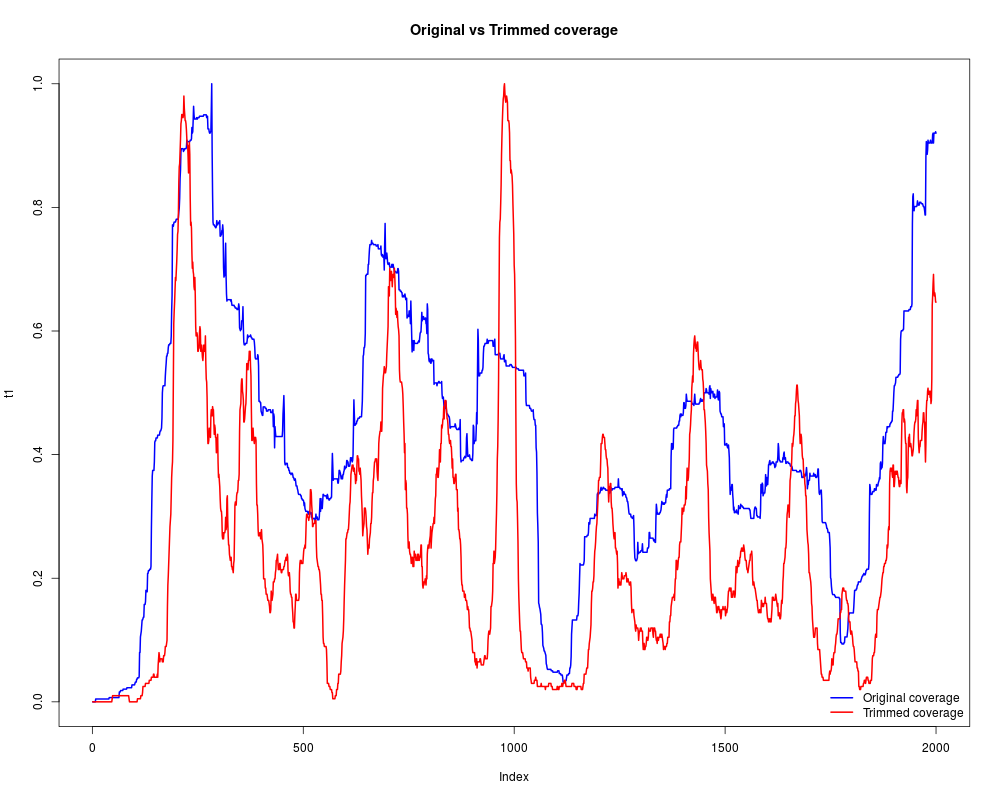
#Compare both coverages, the dyad is much more clear in trimmed version
t1 = as.vector(cover_orig[[1]])[1:2000]
t2 = as.vector(cover_trim[[1]])[1:2000]
t1 = (t1-min(t1))/max(t1-min(t1)) #Normalization
t2 = (t2-min(t2))/max(t2-min(t2)) #Normalization
plot(t1, type="l", lwd="2", col="blue", main="Original vs Trimmed coverage")
lines(t2, lwd="2", col="red")
legend("bottomright", c("Original coverage", "Trimmed coverage"), lwd=2, col=c("blue","red"), bty="n")
#Let's try to call nucleosomes from the trimmed version
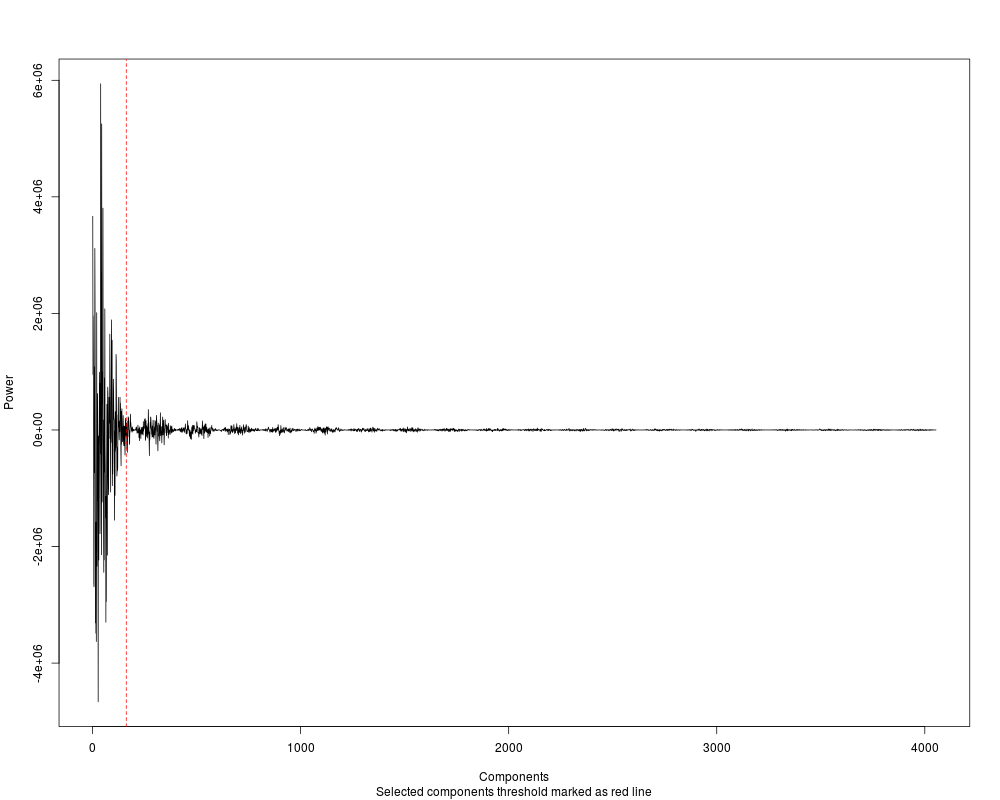
#First of all, let's remove some noise with FFT
#Power spectrum will be plotted, look how with a 2%
#of the components we capture almost all the signal
cover_clean = filterFFT(cover_trim, pcKeepComp=0.02, showPowerSpec=TRUE)
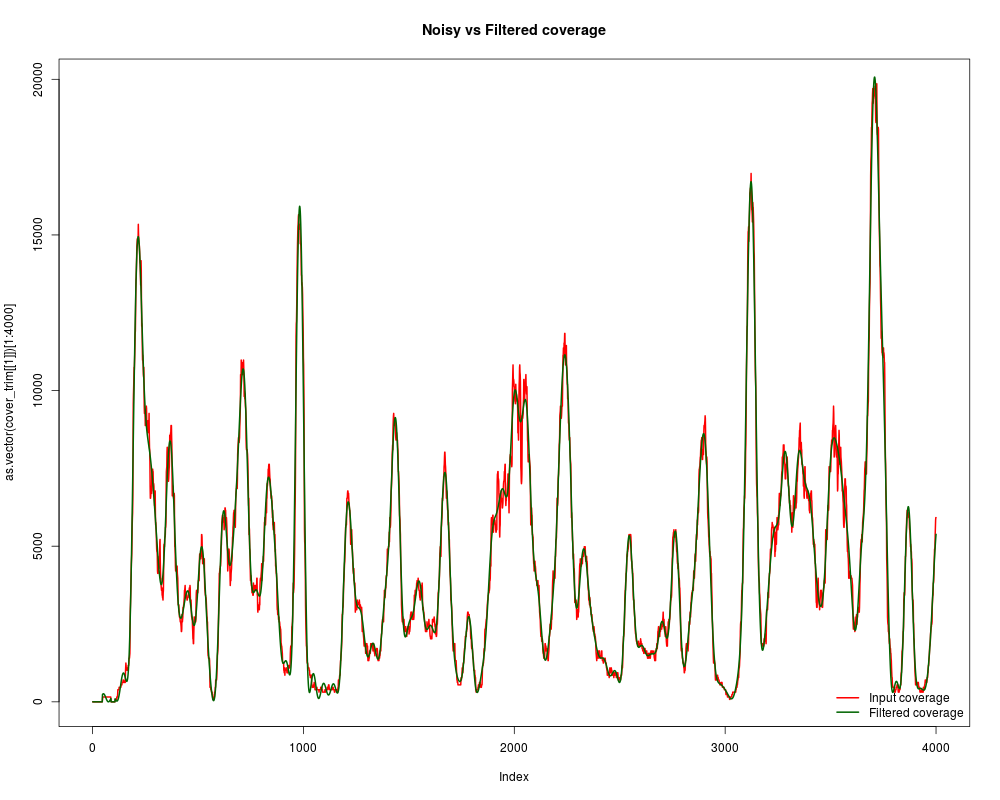
#How clean is now?
plot(as.vector(cover_trim[[1]])[1:4000], t="l", lwd=2, col="red", main="Noisy vs Filtered coverage")
lines(cover_clean[[1]][1:4000], lwd=2, col="darkgreen")
legend("bottomright", c("Input coverage", "Filtered coverage"), lwd=2, col=c("red","darkgreen"), bty="n")
#And how similar? Let's see the correlation
cor(cover_clean[[1]], as.vector(cover_trim[[1]]))
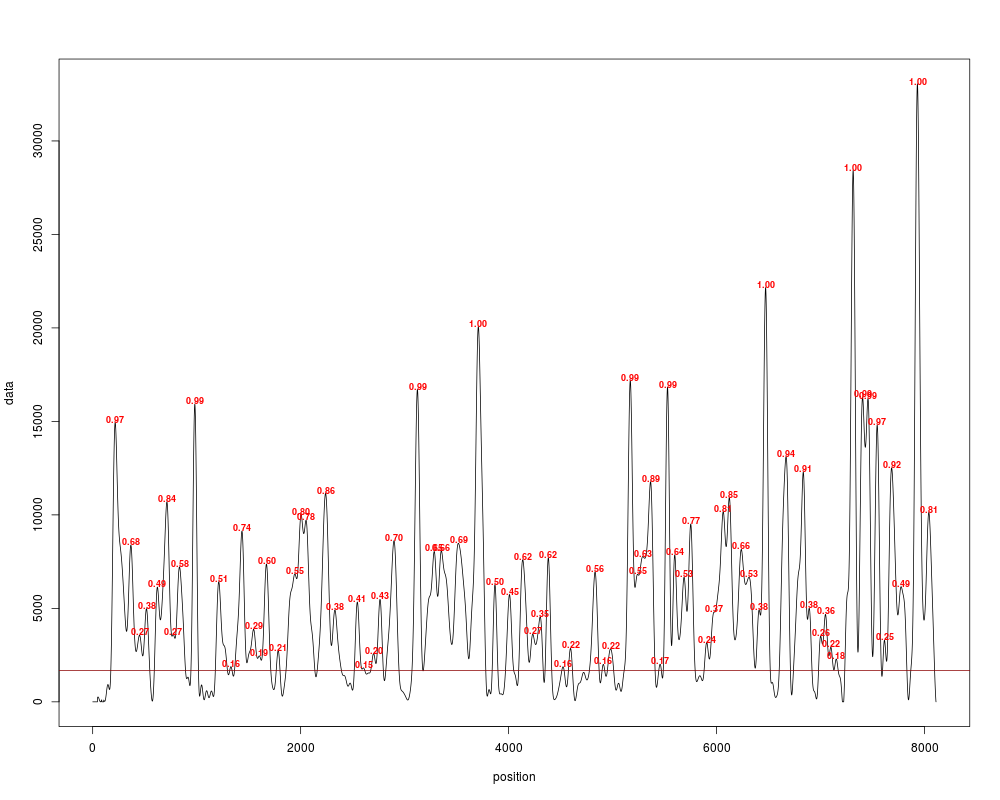
#Now it's time to call for peaks, first just as points
#See that the score is only a measure of the height of the peak
peaks = peakDetection(cover_clean, threshold="25%", score=TRUE)
plotPeaks(peaks[[1]], cover_clean[[1]], threshold="25%")
#Do the same as previously, but now we will create the nucleosome calls:
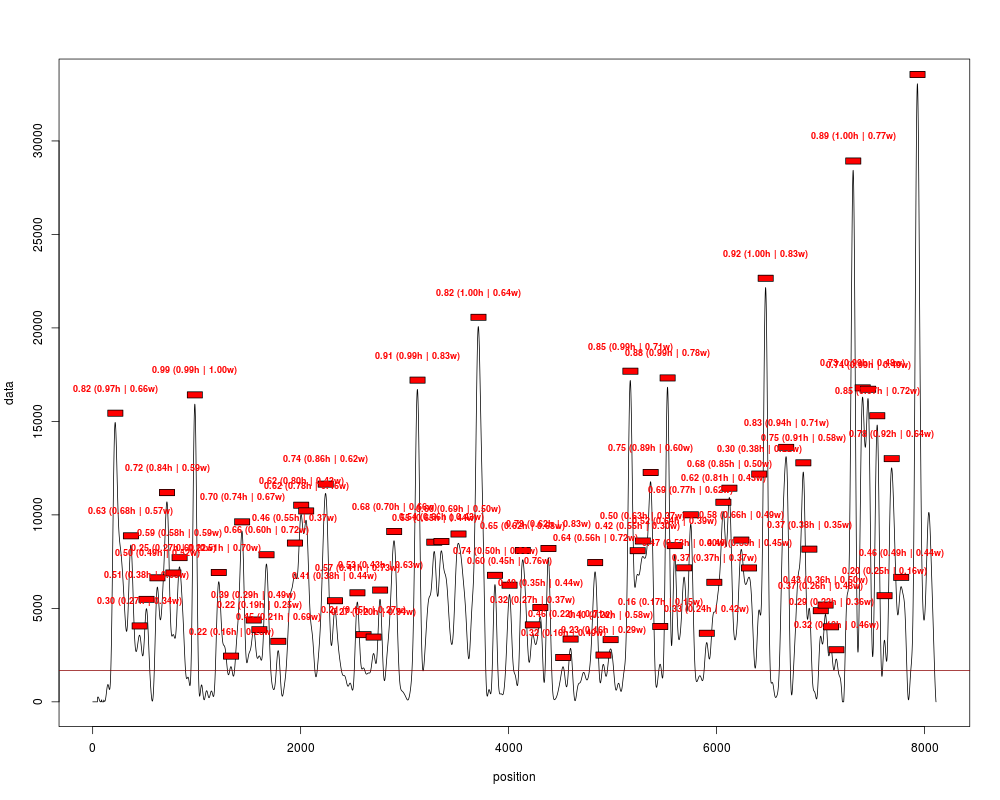
peaks = peakDetection(cover_clean, width=147, threshold="25%", score=TRUE)
plotPeaks(peaks, cover_clean[[1]], threshold="25%")
#This is all. From here, you can filter, merge or work with the nucleosome
#calls using standard IRanges functions and R/Bioconductor manipulation
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(nucleR)
Loading required package: ShortRead
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: BiocParallel
Loading required package: Biostrings
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: GenomicAlignments
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/nucleR/nucleR-package.Rd_%03d_medium.png", width=480, height=480)
> ### Name: nucleR-package
> ### Title: Nucleosome positioning package for R
> ### Aliases: nucleR-package nucleR
> ### Keywords: package
>
> ### ** Examples
>
>
> #Load example dataset:
> # some NGS paired-end reads, mapped with Bowtie and processed with R
> # it is a RangedData object with the start/end coordinates for each read.
> reads = get(data(nucleosome_htseq))
>
> #Process the paired end reads, but discard those with length > 200
> preads_orig = processReads(reads, type="paired", fragmentLen=200)
>
> #Process the reads, but now trim each read to 40bp around the dyad
> preads_trim = processReads(reads, type="paired", fragmentLen=200, trim=40)
>
> #Calculate the coverage, directly in reads per million (r.p.m)
> cover_orig = coverage.rpm(preads_orig)
> cover_trim = coverage.rpm(preads_trim)
>
> #Compare both coverages, the dyad is much more clear in trimmed version
> t1 = as.vector(cover_orig[[1]])[1:2000]
> t2 = as.vector(cover_trim[[1]])[1:2000]
> t1 = (t1-min(t1))/max(t1-min(t1)) #Normalization
> t2 = (t2-min(t2))/max(t2-min(t2)) #Normalization
> plot(t1, type="l", lwd="2", col="blue", main="Original vs Trimmed coverage")
> lines(t2, lwd="2", col="red")
> legend("bottomright", c("Original coverage", "Trimmed coverage"), lwd=2, col=c("blue","red"), bty="n")
>
> #Let's try to call nucleosomes from the trimmed version
> #First of all, let's remove some noise with FFT
> #Power spectrum will be plotted, look how with a 2%
> #of the components we capture almost all the signal
> cover_clean = filterFFT(cover_trim, pcKeepComp=0.02, showPowerSpec=TRUE)
>
> #How clean is now?
> plot(as.vector(cover_trim[[1]])[1:4000], t="l", lwd=2, col="red", main="Noisy vs Filtered coverage")
> lines(cover_clean[[1]][1:4000], lwd=2, col="darkgreen")
> legend("bottomright", c("Input coverage", "Filtered coverage"), lwd=2, col=c("red","darkgreen"), bty="n")
>
> #And how similar? Let's see the correlation
> cor(cover_clean[[1]], as.vector(cover_trim[[1]]))
[1] 0.9937643
>
> #Now it's time to call for peaks, first just as points
> #See that the score is only a measure of the height of the peak
> peaks = peakDetection(cover_clean, threshold="25%", score=TRUE)
> plotPeaks(peaks[[1]], cover_clean[[1]], threshold="25%")
>
> #Do the same as previously, but now we will create the nucleosome calls:
> peaks = peakDetection(cover_clean, width=147, threshold="25%", score=TRUE)
> plotPeaks(peaks, cover_clean[[1]], threshold="25%")
>
> #This is all. From here, you can filter, merge or work with the nucleosome
> #calls using standard IRanges functions and R/Bioconductor manipulation
>
>
>
>
>
> dev.off()
null device
1
>
|
Created & Maintained by Osamu Ogasawara (osamu.ogasawara@gmail.com) and 