Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Import microbio.me/qiime (QIIME-DB) data packageDescriptionOriginally, this function was for accessing microbiome datasets from the microbio.me/qiime public repository from within R. As you can see by clicking on the above link, the QIIME-DB sever is down indefinitely. However, this function will remain supported here in case the FTP server goes back up, and also for phyloseq users that have downloaded one or more data packages prior to the server going down. Usagemicrobio_me_qiime(zipftp, ext = ".zip", parsef = parse_taxonomy_greengenes, ...) Arguments
ValueA See AlsoSee
Examples
# This should return TRUE on your system if you have internet turned on
# and a standard R installation. Indicates whether this is likely to
# work on your system for a URL or local file, respectively.
capabilities("http/ftp"); capabilities("fifo")
# A working example with a local example file included in phyloseq
zipfile = "study_816_split_library_seqs_and_mapping.zip"
zipfile = system.file("extdata", zipfile, package="phyloseq")
tarfile = "study_816_split_library_seqs_and_mapping.tar.gz"
tarfile = system.file("extdata", tarfile, package="phyloseq")
tarps = microbio_me_qiime(tarfile)
zipps = microbio_me_qiime(zipfile)
identical(tarps, zipps)
tarps; zipps
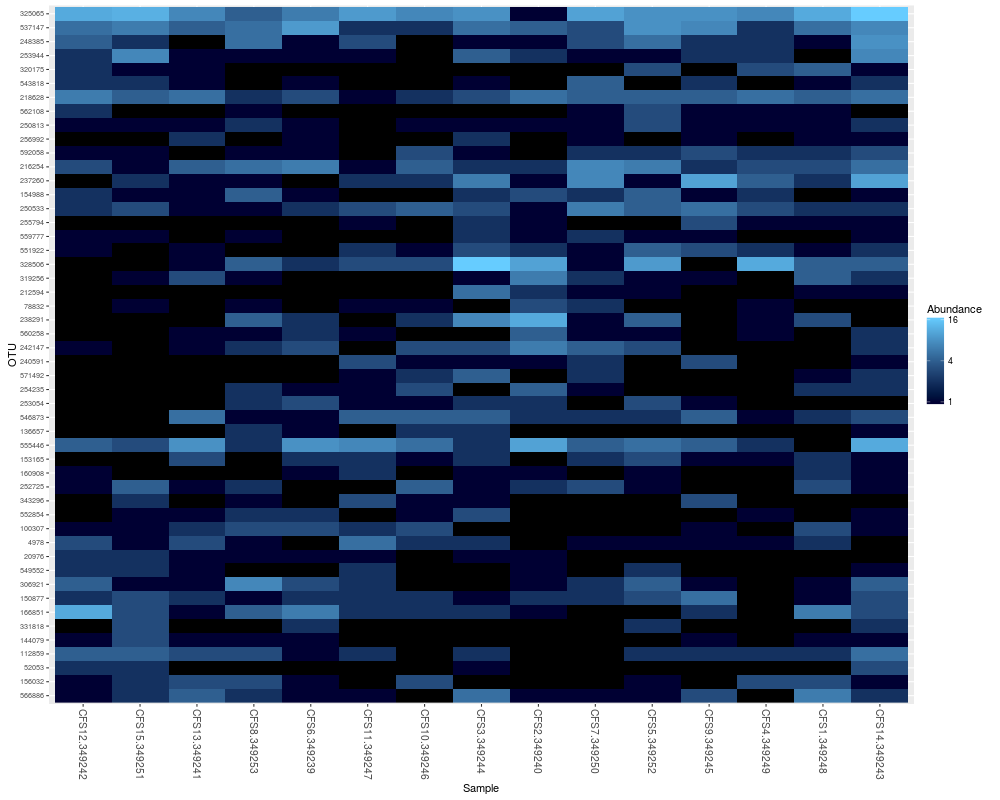
plot_heatmap(tarps)
# An example that used to work, before the QIIME-DB server was turned off by its host.
# # Smokers dataset
# smokezip = "ftp://thebeast.colorado.edu/pub/QIIME_DB_Public_Studies/study_524_split_library_seqs_and_mapping.zip"
# smokers1 = microbio_me_qiime(smokezip)
# # Alternatively, just use the study number
# smokers2 = microbio_me_qiime(524)
# identical(smokers1, smokers2)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(phyloseq)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/phyloseq/microbio_me_qiime.Rd_%03d_medium.png", width=480, height=480)
> ### Name: microbio_me_qiime
> ### Title: Import microbio.me/qiime (QIIME-DB) data package
> ### Aliases: microbio_me_qiime
>
> ### ** Examples
>
> # This should return TRUE on your system if you have internet turned on
> # and a standard R installation. Indicates whether this is likely to
> # work on your system for a URL or local file, respectively.
> capabilities("http/ftp"); capabilities("fifo")
http/ftp
TRUE
fifo
TRUE
> # A working example with a local example file included in phyloseq
> zipfile = "study_816_split_library_seqs_and_mapping.zip"
> zipfile = system.file("extdata", zipfile, package="phyloseq")
> tarfile = "study_816_split_library_seqs_and_mapping.tar.gz"
> tarfile = system.file("extdata", tarfile, package="phyloseq")
> tarps = microbio_me_qiime(tarfile)
Found biom-format file, now parsing it...
Done parsing biom...
Importing Sample Metdadata from mapping file...
Merging the imported objects...
Successfully merged, phyloseq-class created.
Returning...
Warning message:
In microbio_me_qiime(tarfile) : NAs introduced by coercion
> zipps = microbio_me_qiime(zipfile)
Found biom-format file, now parsing it...
Done parsing biom...
Importing Sample Metdadata from mapping file...
Merging the imported objects...
Successfully merged, phyloseq-class created.
Returning...
Warning message:
In microbio_me_qiime(zipfile) : NAs introduced by coercion
> identical(tarps, zipps)
[1] TRUE
> tarps; zipps
phyloseq-class experiment-level object
otu_table() OTU Table: [ 50 taxa and 15 samples ]
sample_data() Sample Data: [ 15 samples by 39 sample variables ]
tax_table() Taxonomy Table: [ 50 taxa by 7 taxonomic ranks ]
phyloseq-class experiment-level object
otu_table() OTU Table: [ 50 taxa and 15 samples ]
sample_data() Sample Data: [ 15 samples by 39 sample variables ]
tax_table() Taxonomy Table: [ 50 taxa by 7 taxonomic ranks ]
> plot_heatmap(tarps)
> # An example that used to work, before the QIIME-DB server was turned off by its host.
> # # Smokers dataset
> # smokezip = "ftp://thebeast.colorado.edu/pub/QIIME_DB_Public_Studies/study_524_split_library_seqs_and_mapping.zip"
> # smokers1 = microbio_me_qiime(smokezip)
> # # Alternatively, just use the study number
> # smokers2 = microbio_me_qiime(524)
> # identical(smokers1, smokers2)
>
>
>
>
>
> dev.off()
null device
1
>
|