Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
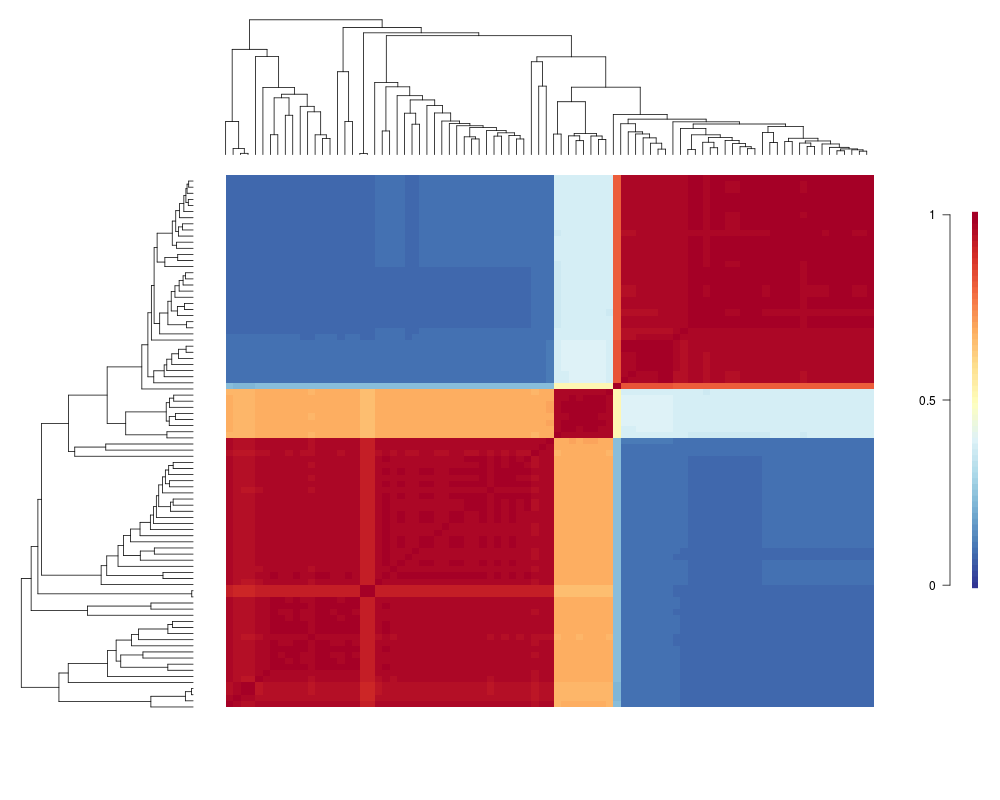
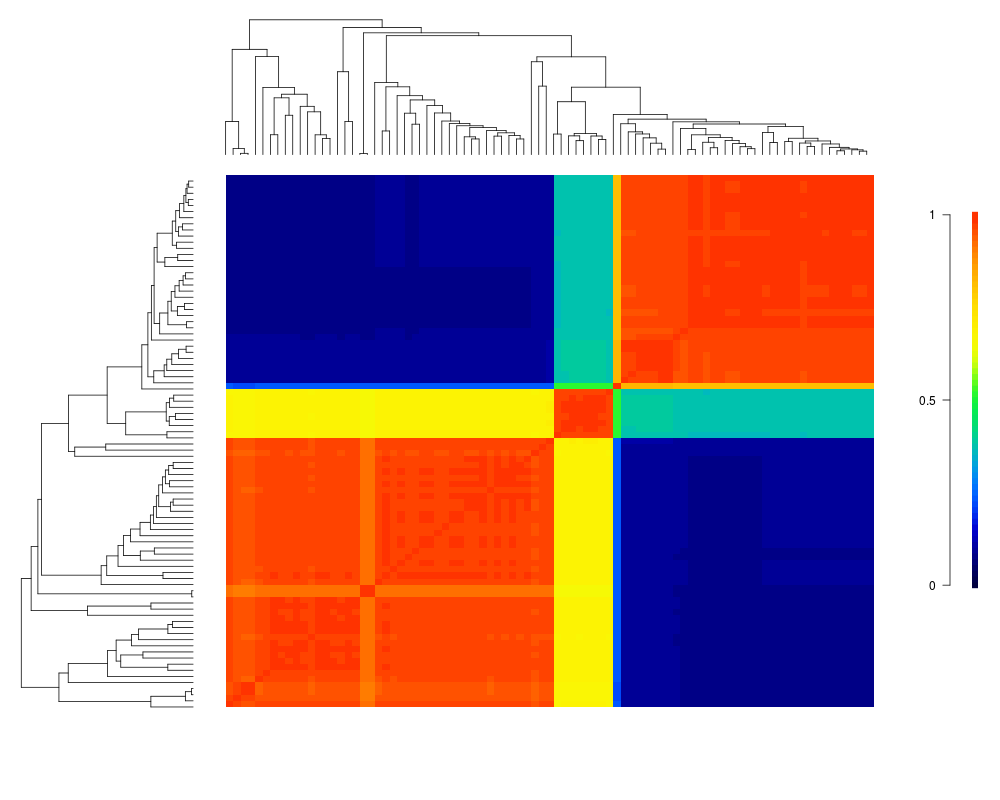
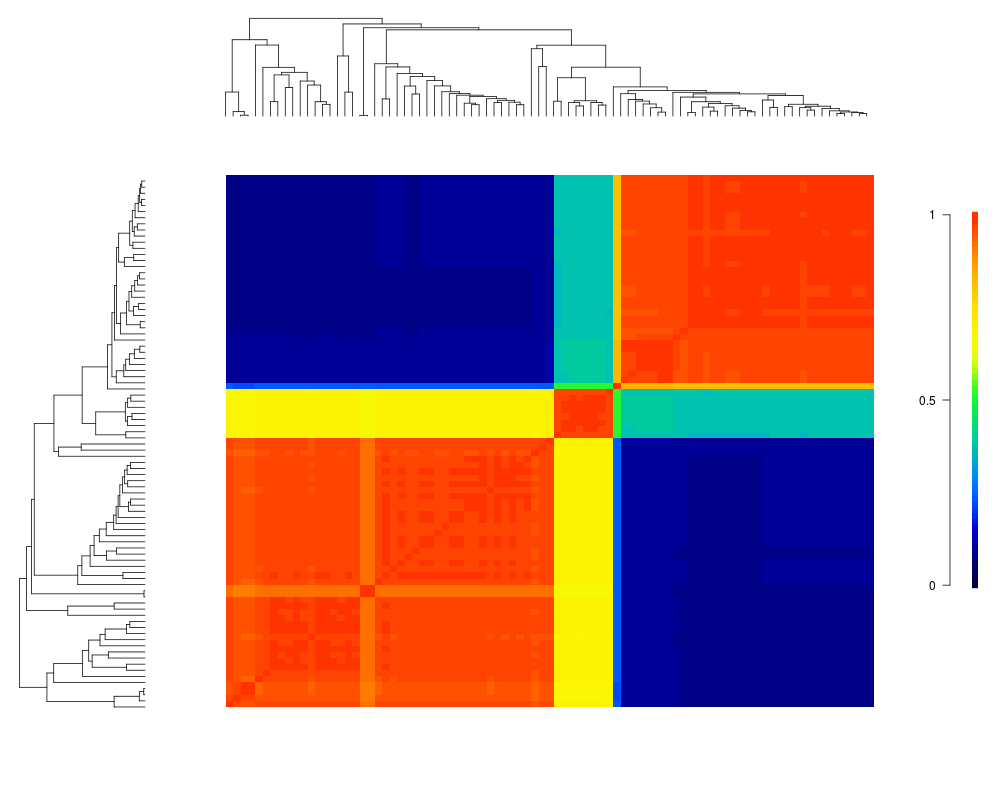
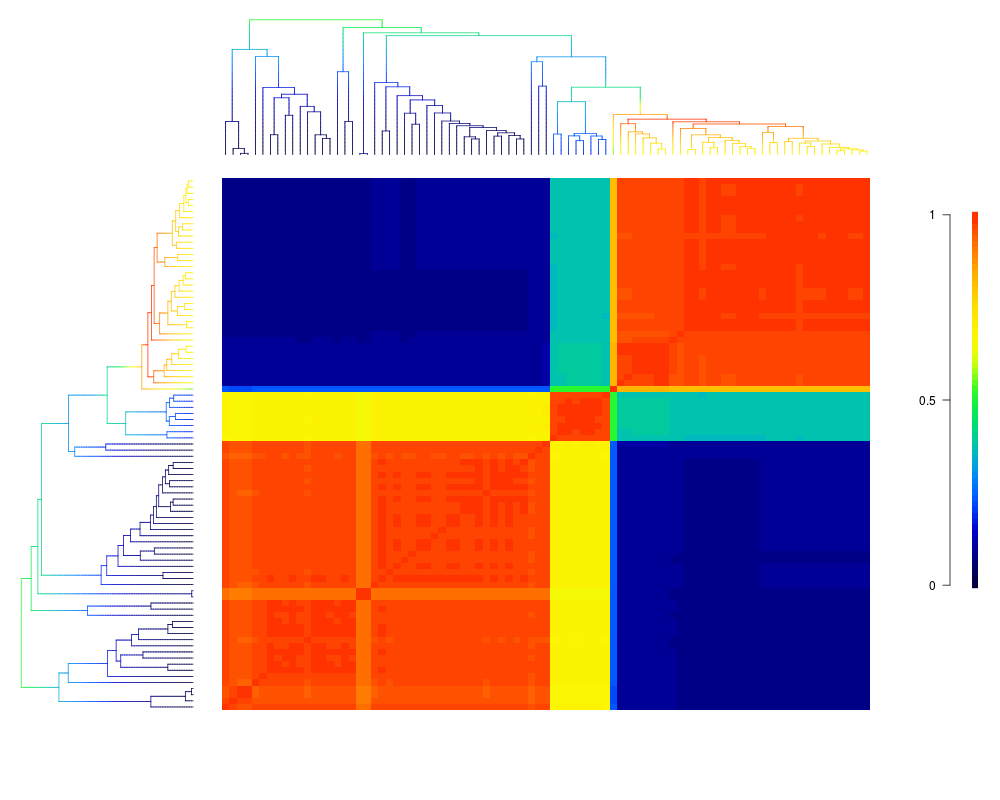
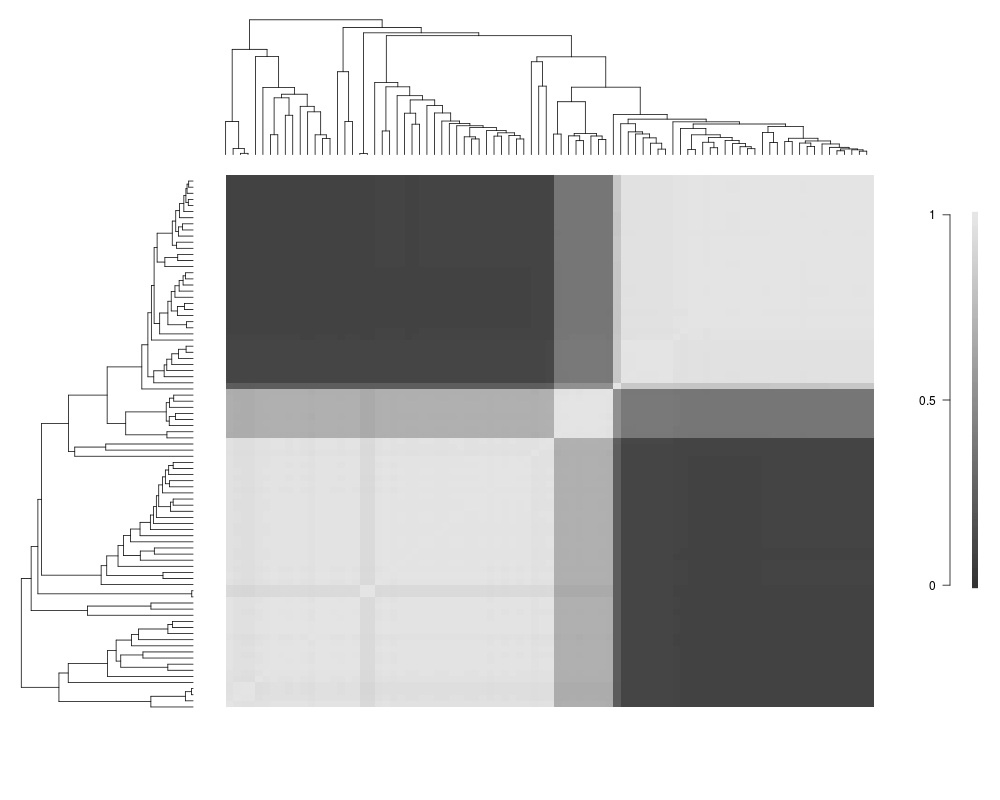
Visualize macroevolutionary cohortsDescriptionPlots the matrix of pairwise correlations in rate regimes between all tips in a phylogeny. Usagecohorts(x, ephy, col, pal, lwd = 1, ofs = 0, use.plot.bammdata = FALSE, useraster = FALSE, LARGE = 500, ...) Arguments
DetailsThe plotting function creates an image of the IMPORTANT: the legend DOES NOT apply to the phylorate plots
shown in the margin if Author(s)Mike Grundler See Also
Examples
data(whales, events.whales)
ed <- getEventData(whales, events.whales, burnin=0.1, nsamples=500)
x <- getCohortMatrix(ed)
cohorts(x, ed)
cohorts(x, ed, col='temperature')
cohorts(x, ed, ofs=0.05, col='temperature')
cohorts(x, ed, pal="temperature", col='temperature', use.plot.bammdata=TRUE)
# gray scale
cohorts(x, ed, col=gray(seq(0.2,0.9,length.out=128)),
use.plot.bammdata=FALSE)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(BAMMtools)
Loading required package: ape
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/BAMMtools/cohorts.Rd_%03d_medium.png", width=480, height=480)
> ### Name: cohorts
> ### Title: Visualize macroevolutionary cohorts
> ### Aliases: cohorts
>
> ### ** Examples
>
> data(whales, events.whales)
> ed <- getEventData(whales, events.whales, burnin=0.1, nsamples=500)
Processing event data from data.frame
Discarded as burnin: GENERATIONS < 995000
Analyzing 500 samples from posterior
Setting recursive sequence on tree...
Done with recursive sequence
> x <- getCohortMatrix(ed)
> cohorts(x, ed)
> cohorts(x, ed, col='temperature')
> cohorts(x, ed, ofs=0.05, col='temperature')
> cohorts(x, ed, pal="temperature", col='temperature', use.plot.bammdata=TRUE)
> # gray scale
> cohorts(x, ed, col=gray(seq(0.2,0.9,length.out=128)),
+ use.plot.bammdata=FALSE)
>
>
>
>
>
> dev.off()
null device
1
>
|