Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Plot
|
x |
An object of class |
tau |
A numeric indicating the grain size for the calculations. See
documentation for |
method |
A character string indicating the method for plotting the
phylogenetic tree. |
xlim |
A numeric vector of coordinates for the x-axis endpoints.
Defaults to |
ylim |
A numeric vector of coordinates for the y-axis endpoints.
Defaults to |
vtheta |
A numeric indicating the angular separation (in degrees) of
the first and last terminal nodes. Ignored if
|
rbf |
A numeric indicating the length of the root branch as a
fraction of total tree height. Ignored if |
show |
A logical indicating whether or not to plot the tree. Defaults
to |
labels |
A logical indicating whether or not to plot the tip labels.
Defaults to |
legend |
A logical indicating whether or not to plot a legend for
interpreting the mapping of evolutionary rates to colors. Defaults to
|
spex |
A character string indicating what type of macroevolutionary
rates should be plotted. "s" (default) indicates speciation rates, "e"
indicates extinction rates, and "netdiv" indicates net diversification
rates. Ignored if |
lwd |
A numeric specifying the line width for branches. |
cex |
A numeric specifying the size of tip labels. |
pal |
A character string or vector of mode character that describes the color palette. See Details for explanation of options. |
mask |
An optional integer vector of node numbers specifying branches
that will be masked with |
mask.color |
The color for the mask. |
colorbreaks |
A numeric vector of percentiles delimiting the bins for
mapping rates to colors. If |
logcolor |
Logical. Should colors be plotted on a log scale. |
breaksmethod |
Method used for determining color breaks. See help
file for |
color.interval |
Min and max value for the mapping of rates. If
|
JenksSubset |
If |
par.reset |
A logical indicating whether or not to reset the
graphical parameters when the function exits. Defaults to
|
direction |
A character string. Options are "rightwards", "leftwards", "upwards", and "downwards", which determine the orientation of the tips when the phylogeny plotted. |
... |
Further arguments passed to |
Details
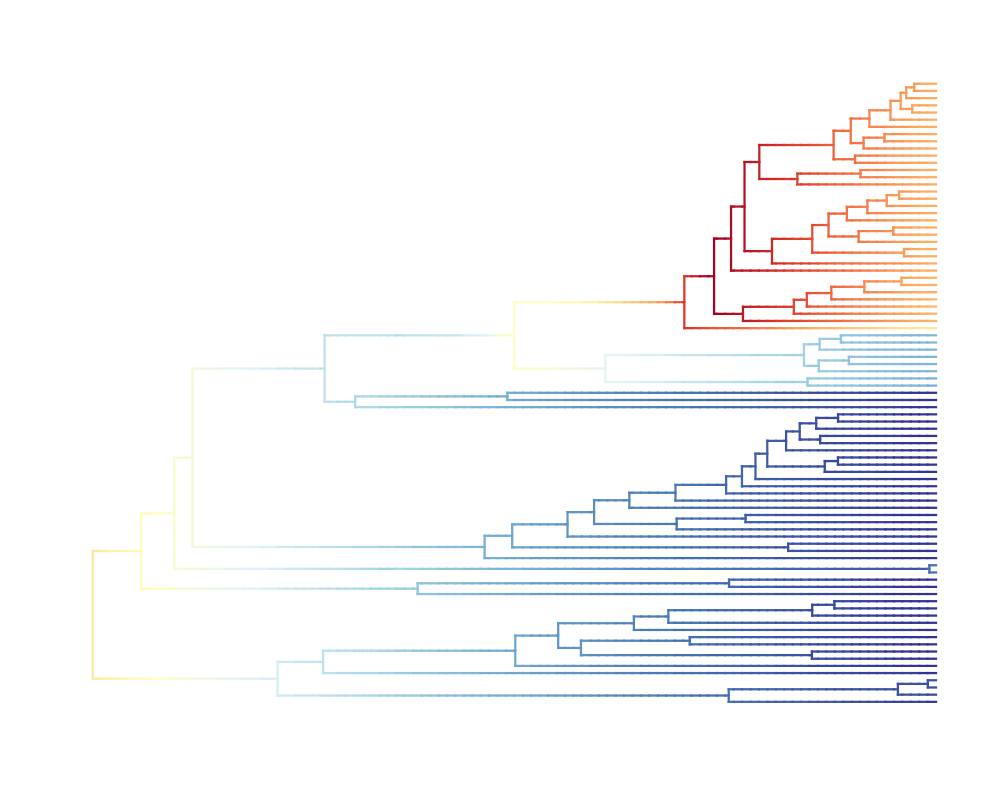
To calculate rates, each branch of the phylogeny is discretized
into a number of small segments, and the mean of the marginal
posterior density of the rate of speciation/extinction or trait
evolution is calculated for each such segment. Rates are mapped to
colors such that cool colors represent slow rates and warm colors
represent fast rates. When the tree is plotted each of these small
segments is plotted so that changes in rates through time and shifts
in rates are visible as gradients of color. The spex argument
determines the type of rate that will be calculated. spex = "s"
will plot speciation rates, spex = "e" will plot extinction
rates, and spex = "netdiv" will plot diversification rates
(speciation - extinction). Note that if x$type = "trait" the
spex argument is ignored and rates of phenotypic evolution are
plotted instead. If legend = TRUE the function will plot a
legend that contains the mapping of colors to numerical values.
A number of color palettes come built in with BAMMtools.
Color-blind friendly options include:
BrBG
PiYG
PRGn
PuOr
RdBu
RdYlBu
BuOr
BuOrRd
DkRdBu
BuDkOr
GnPu
Some color-blind unfriendly options include:
RdYlGn
Spectral
temperature
terrain
Some grayscale options include:
grayscale
revgray
For more information about these color palettes visit
http://colorbrewer2.org and
http://geography.uoregon.edu/datagraphics/color_scales.htm or
use the help files of the R packages RColorBrewer and
dichromat.
Additionally, any vector of valid named colors may also be used. The
only restriction is that the length of this vector be greater than or
equal to three (you can provide a single color, but in this case the
entire tree will be assigned the same color). The colors should be
ordered from cool to warm as the colors will be mapped from low rates
to high rates in the order supplied (e.g. pal=c("darkgreen",
"yellow2", "red")). The option pal = "temperature" uses the
rich.colors function written by Arni Magnusson for the R
package gplots.
Internally plot.bammdata checks whether or not rates have been
calculated by looking for a component named "dtrates" in the
bammdata object. If rates have not been calculated
plot.bammdata calls dtRates with tau. Specifying
smaller values for tau will result in smoother-looking rate
changes on the tree. Note that smaller values of tau require
more computation. If the colorbreaks argument
is NULL a map of rates to colors is also made by calling
assignColorBreaks with NCOLORS = 64. A user supplied
colorbreaks argument can be passed as well. This allows one to
plot parts of a tree while preserving the map of rates to colors that
was made using rates for the entire tree.
If color.interval is defined, then those min and max values override the automatic detection of min and max. This might be useful if some small number of lineages have very high or very low rates, such that the map of colors is being skewed towards these extremes, resulting in other rate variation being drowned out. If specified, the color ramp will be built between these two color.interval values, and the rates outside of the color interval range will be set to the highest and lowest color.
If plot.bammdata is called repeatedly with the same
bammdata object, computation can be reduced by first calling
dtRates in the global environment.
Value
Returns (invisibly) a list with three components.
coords A matrix of plot coordinates. Rows correspond to branches. Columns 1-2 are starting (x,y) coordinates of each branch and columns 3-4 are ending (x,y) coordinates of each branch. If
method = "polar"a fifth column gives the angle(in radians) of each branch.colorbreaks A vector of percentiles used to group macroevolutionary rates into color bins.
colordens A matrix of the kernel density estimates (column 2) of evolutionary rates (column 1) and the color (column 3) corresponding to each rate value.
Author(s)
Mike Grundler, Pascal Title
Source
http://colorbrewer2.org, http://geography.uoregon.edu/datagraphics/color_scales.htm
See Also
dtRates, addBAMMshifts,
assignColorBreaks, subtreeBAMM,
colorRampPalette
Examples
data(whales, events.whales)
ed <- getEventData(whales, events.whales, burnin=0.25, nsamples=500)
# The first call to plot.bammdata
# No calculations or assignments of rates have been made
plot(ed, lwd = 3, spex = "s") # calls dtRates & assignColorBreaks
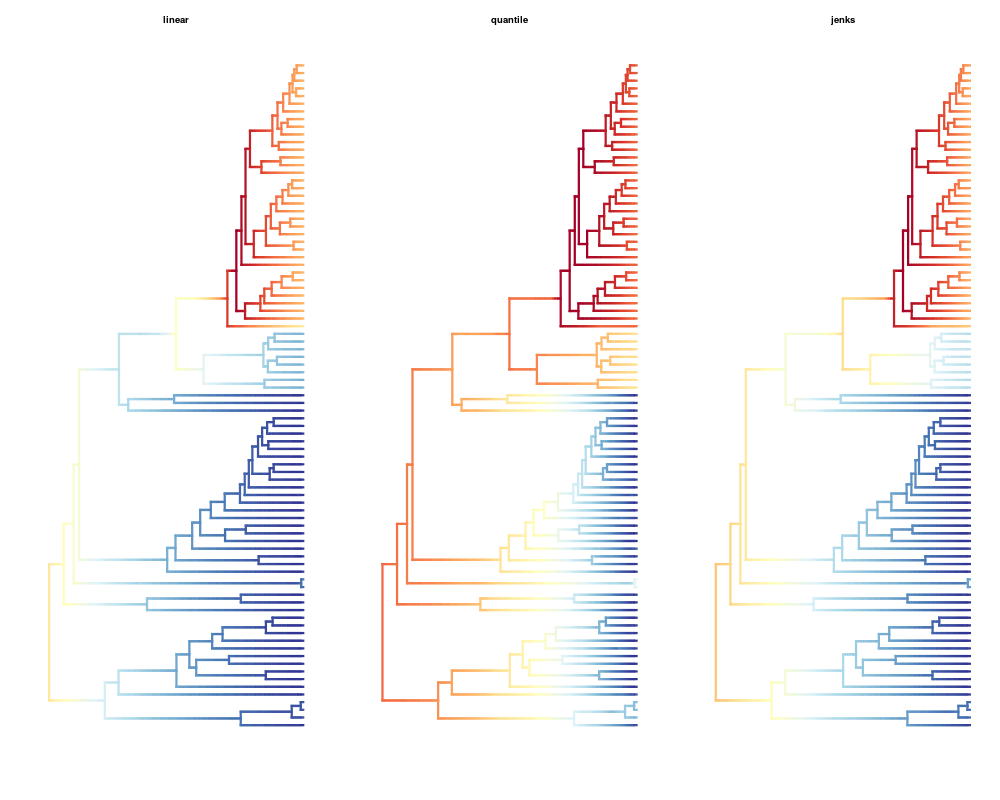
# Compare the different color breaks methods
par(mfrow=c(1,3))
plot(ed, lwd = 3, spex = "s", breaksmethod = "linear")
title(main="linear")
plot(ed, lwd = 3, spex = "s", breaksmethod = "quantile")
title(main="quantile")
plot(ed, lwd = 3, spex = "s", breaksmethod = "jenks")
title(main="jenks")
## Not run:
# now plot.bammdata no longer calls dtRates
ed <- dtRates(ed, tau = 0.01)
xx <- plot(ed, lwd = 3, spex = "s")
# you can plot subtrees while preserving the original
# rates to colors map by passing the colorbreaks object as an argument
sed <- subtreeBAMM(ed, node = 103)
plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
sed <- subtreeBAMM(ed, node = 140)
plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
# note how if we do not pass colorbreaks the map is
# no longer relative to the rest of the tree and the plot is quite
# distinct from the original
plot(sed, lwd = 3)
# if you want to change the value of tau and the rates to colors map for
# the entire tree
ed <- dtRates(ed, tau = 0.002)
xx <- plot(ed, lwd = 3, spex = "s")
# now you can re-plot the subtrees using this finer tau partition
sed <- subtreeBAMM(ed, node = 103)
sed <- dtRates(sed, 0.002)
plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
sed <- subtreeBAMM(ed, node = 140)
sed <- dtRates(sed, 0.002)
plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
# multi-panel plotting and adding shifts of specific posterior samples
par(mfrow=c(2,3))
samples <- sample(1:length(ed$eventData), 6)
ed <- dtRates(ed, 0.005)
# individual plots will have a color map relative to the mean
xx <- plot(ed, show=FALSE)
for (i in 1:6) {
ed <- dtRates(ed, 0.005, samples[i])
plot(ed, colorbreaks=xx$colorbreaks)
addBAMMshifts(ed,index=samples[i],method="phylogram", par.reset=FALSE)
}
dev.off()
# color options
ed <- dtRates(ed,0.01)
plot(ed, pal="temperature",lwd=3)
plot(ed, pal="terrain",lwd=3)
plot(ed, pal=c("darkgreen","yellow2","red"),lwd=3)
plot(ed,method="polar",pal="Spectral", lwd=3)
plot(ed,method="polar",pal="RdYlBu", lwd=3)
## End(Not run)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(BAMMtools)
Loading required package: ape
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/BAMMtools/plot.bammdata.Rd_%03d_medium.png", width=480, height=480)
> ### Name: plot.bammdata
> ### Title: Plot 'BAMM'-estimated macroevolutionary rates on a phylogeny
> ### Aliases: plot.bammdata
> ### Keywords: graphics models
>
> ### ** Examples
>
> data(whales, events.whales)
> ed <- getEventData(whales, events.whales, burnin=0.25, nsamples=500)
Processing event data from data.frame
Discarded as burnin: GENERATIONS < 2495000
Analyzing 500 samples from posterior
Setting recursive sequence on tree...
Done with recursive sequence
>
> # The first call to plot.bammdata
> # No calculations or assignments of rates have been made
> plot(ed, lwd = 3, spex = "s") # calls dtRates & assignColorBreaks
>
> # Compare the different color breaks methods
> par(mfrow=c(1,3))
> plot(ed, lwd = 3, spex = "s", breaksmethod = "linear")
> title(main="linear")
> plot(ed, lwd = 3, spex = "s", breaksmethod = "quantile")
> title(main="quantile")
> plot(ed, lwd = 3, spex = "s", breaksmethod = "jenks")
> title(main="jenks")
>
> ## Not run:
> ##D # now plot.bammdata no longer calls dtRates
> ##D ed <- dtRates(ed, tau = 0.01)
> ##D xx <- plot(ed, lwd = 3, spex = "s")
> ##D
> ##D # you can plot subtrees while preserving the original
> ##D # rates to colors map by passing the colorbreaks object as an argument
> ##D sed <- subtreeBAMM(ed, node = 103)
> ##D plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
> ##D sed <- subtreeBAMM(ed, node = 140)
> ##D plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
> ##D # note how if we do not pass colorbreaks the map is
> ##D # no longer relative to the rest of the tree and the plot is quite
> ##D # distinct from the original
> ##D plot(sed, lwd = 3)
> ##D
> ##D # if you want to change the value of tau and the rates to colors map for
> ##D # the entire tree
> ##D ed <- dtRates(ed, tau = 0.002)
> ##D xx <- plot(ed, lwd = 3, spex = "s")
> ##D # now you can re-plot the subtrees using this finer tau partition
> ##D sed <- subtreeBAMM(ed, node = 103)
> ##D sed <- dtRates(sed, 0.002)
> ##D plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
> ##D sed <- subtreeBAMM(ed, node = 140)
> ##D sed <- dtRates(sed, 0.002)
> ##D plot(sed, lwd = 3, colorbreaks = xx$colorbreaks)
> ##D
> ##D # multi-panel plotting and adding shifts of specific posterior samples
> ##D par(mfrow=c(2,3))
> ##D samples <- sample(1:length(ed$eventData), 6)
> ##D ed <- dtRates(ed, 0.005)
> ##D # individual plots will have a color map relative to the mean
> ##D xx <- plot(ed, show=FALSE)
> ##D for (i in 1:6) {
> ##D ed <- dtRates(ed, 0.005, samples[i])
> ##D plot(ed, colorbreaks=xx$colorbreaks)
> ##D addBAMMshifts(ed,index=samples[i],method="phylogram", par.reset=FALSE)
> ##D }
> ##D dev.off()
> ##D
> ##D # color options
> ##D ed <- dtRates(ed,0.01)
> ##D plot(ed, pal="temperature",lwd=3)
> ##D plot(ed, pal="terrain",lwd=3)
> ##D plot(ed, pal=c("darkgreen","yellow2","red"),lwd=3)
> ##D plot(ed,method="polar",pal="Spectral", lwd=3)
> ##D plot(ed,method="polar",pal="RdYlBu", lwd=3)
> ## End(Not run)
>
>
>
>
>
> dev.off()
null device
1
>
|