Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Bayesian Composition EstimatorDescriptionThis function estimates taxonomic compositions of algal communities
based on biomarker field data. More precisely, it estimates the probability distributions of a sample composition
based on an input ratio matrix, Probability distributions are estimated based on an adaptive metropolis
MCMC method, function Usagebce1(A, B, Wa=NULL, Wb=NULL,
jmpType="default", jmpA=.1,jmpX=.1, jmpCovar=NULL,
initX=NULL, initA=NULL, priorA="normal", minA=NULL, maxA=NULL,
var0=NULL, wvar0=1e-6, Xratios=TRUE, verbose=TRUE, ...)
Arguments
DetailsThe function A%*%X ~= B It does this by returning data matrix B:
and ratio matrix A:
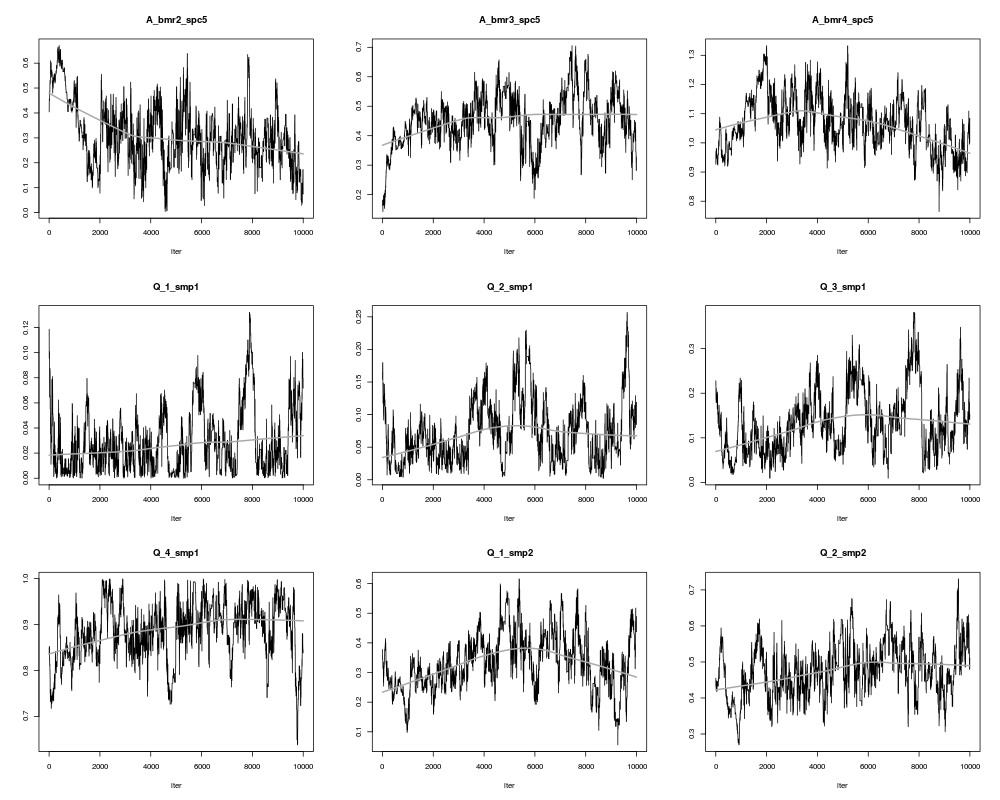
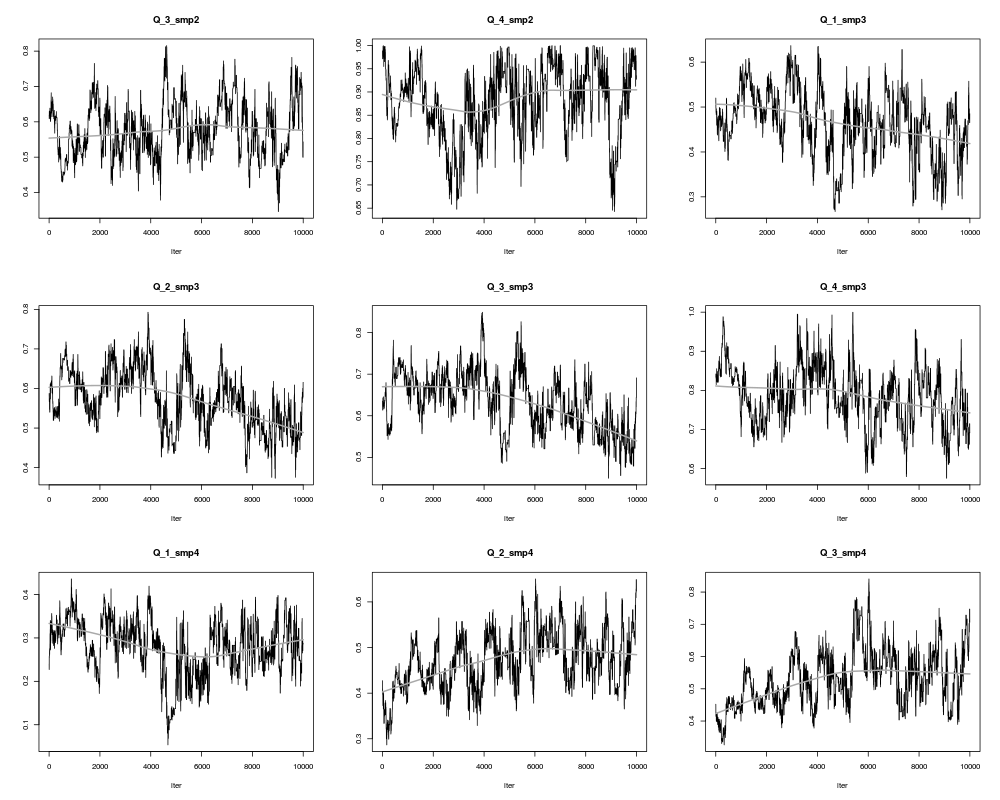
ValueAn object of class bce and _modMCMC_ (returned by the function modMCMC). This object has methods for the generic functions 'summary', 'plot', 'pairs'- see ?modMCMC. It is distinguished from other modMCMC objects by 3 extra attributes that allow to extract matrices A and X from the mcmc result: "dim_A" (dimensions of A), "A_not_null" (which elements of A are not zero and thus included in the mcmc) and Xratios (whether X was rescaled, yes or no). NoteProducing sensible output: Markov Chain Monte Carlo simulations are not as straightforward as one might wish; several preliminary runs might be necessary to determine the desired number of iterations, burn-in length and jump length. For all estimated values of Rat and X, their trace (evolution of the values over all iterations) has to display random behaviour; no obvious trends should appear. A few parameters can be tuned to obtain such behaviour:
Author(s)Karel Van den Meersche <k.vdmeersche@nioo.knaw.nl>, Karline Soetaert <k.soetaert@nioo.knaw.nl>. ReferencesVan den Meersche, K., K. Soetaert and J.J. Middelburg (2008) A Bayesian compositional estimator for microbial taxonomy based on biomarkers, Limnology and Oceanography Methods 6, 190-199 See Also
Examples
##====================================
# example using bceInput data
# !!! should be weighted to correspond better to example of BCE!!!
A <- t(bceInput$Rat)
B <- t(bceInput$Dat)
result <- bce1(A,B,niter=1000)
## the number of accepted runs is zero;
## try different starting values
result <- bce1(A,B,niter=1000,initX=matrix(1/ncol(A),ncol(A),ncol(B)))
## number of accepted runs is still low;
## smaller jumps
result <- bce1(A,B,niter=1000,initX=matrix(1/ncol(A),ncol(A),ncol(B)),jmpA=.01,jmpX=.01)
Sum <-summary(result)
## did the algorithm converge?
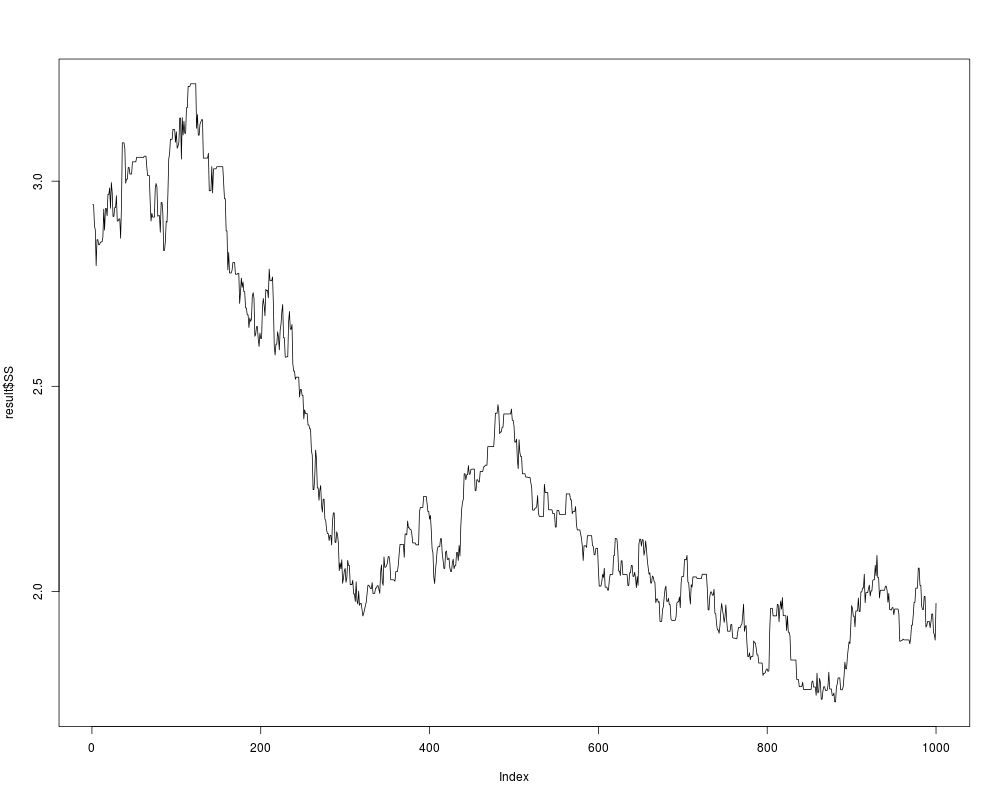
plot(result$SS,type="l")
## no
## more runs, using the output of previous run as input.
result <- bce1(A,B,niter=1e4,jmpA=.01,jmpX=.01,updatecov=1e3,
initX=Sum$lastX,initA=Sum$lastA,
jmpCovar=Sum$covar*2.4^2/ncol(result$pars),
)
Sum <-summary(result)
## we inspect the output:
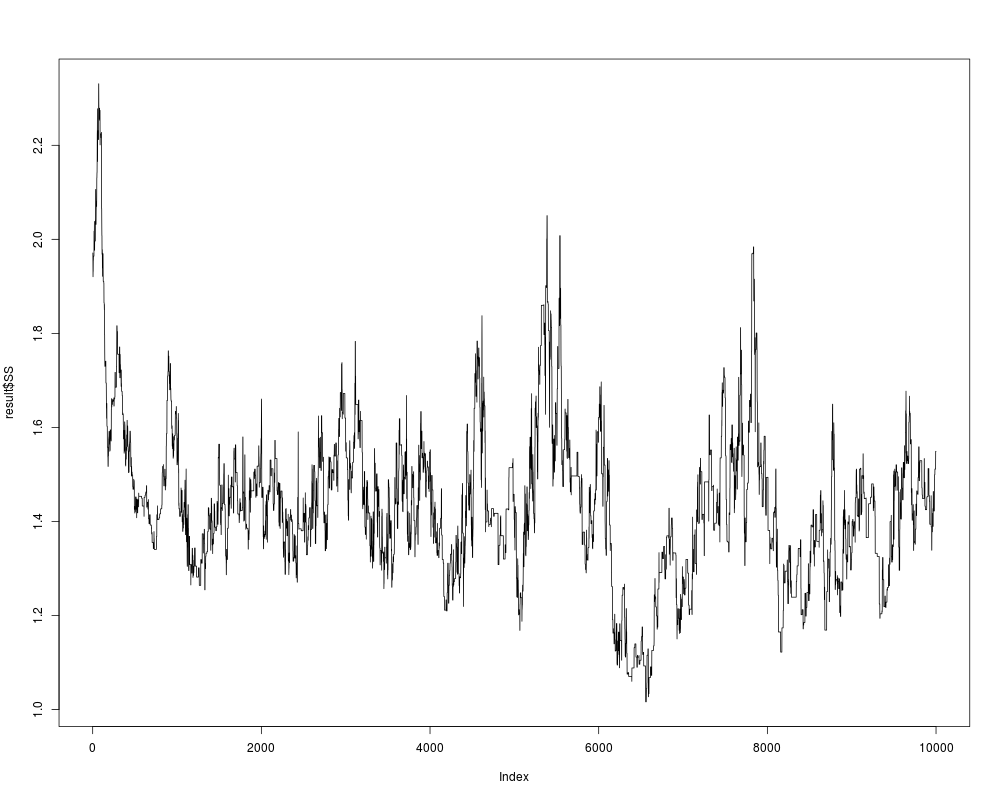
plot(result$SS,type="l")
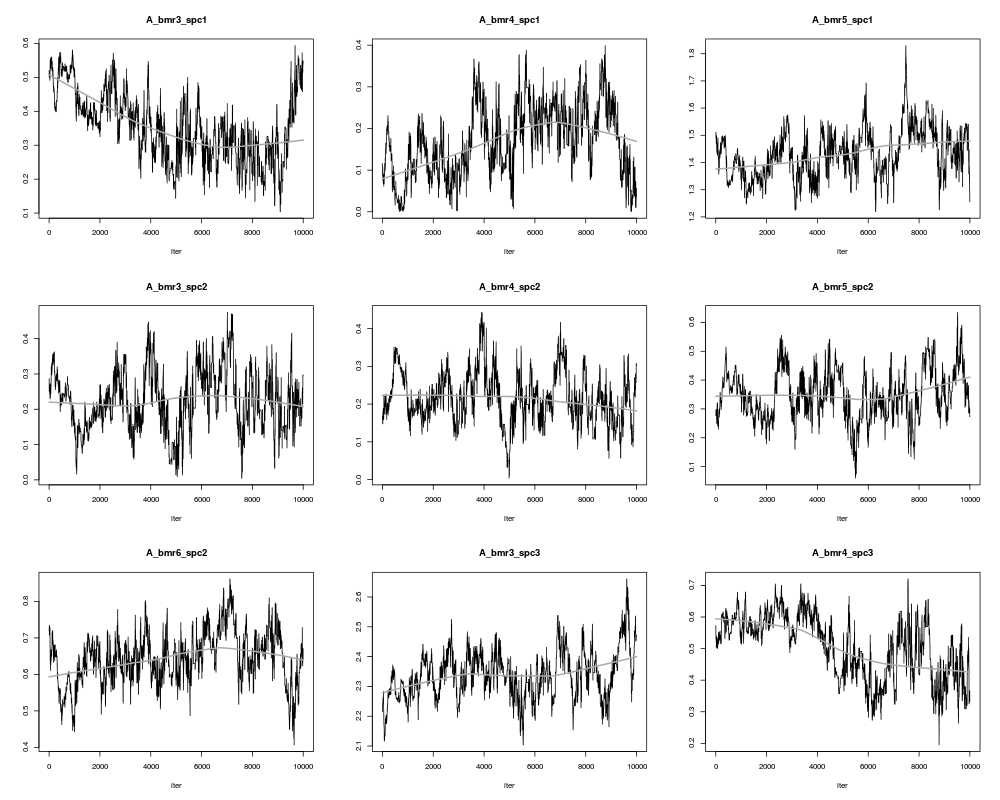
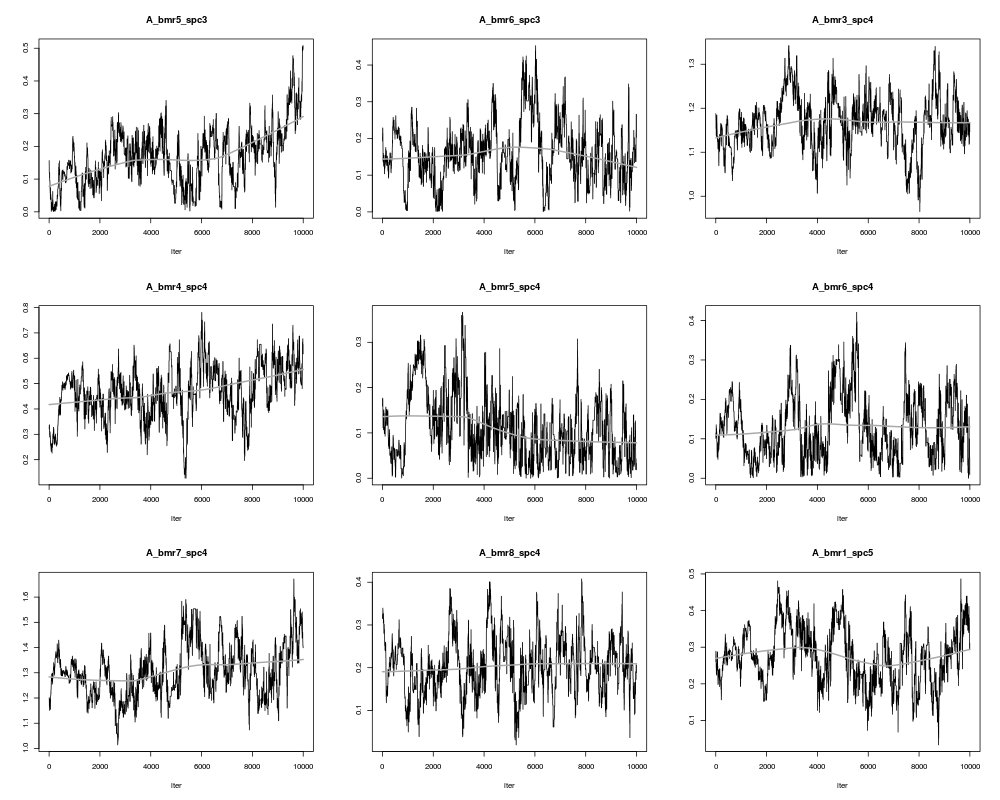
plot(result,ask=TRUE)
## looks already pretty good; to get a better result, repeat one more
## time with a longer run. Uncomment the following paragraph and run.
## go get some coffee, this might take a while (~30s).
## result <- bce1(A,B,niter=1e5,jmpA=.01,jmpX=.01,updatecov=1e3,
## outputlength=1e3,burninlength=.35e5,
## initX=Sum$lastX,initA=Sum$lastA,
## jmpCovar=Sum$covar*2.4^2/ncol(result$pars),
## )
## Sum <-summary(result)
## plot(result$SS,type="l")
## plot(result,ask=TRUE)
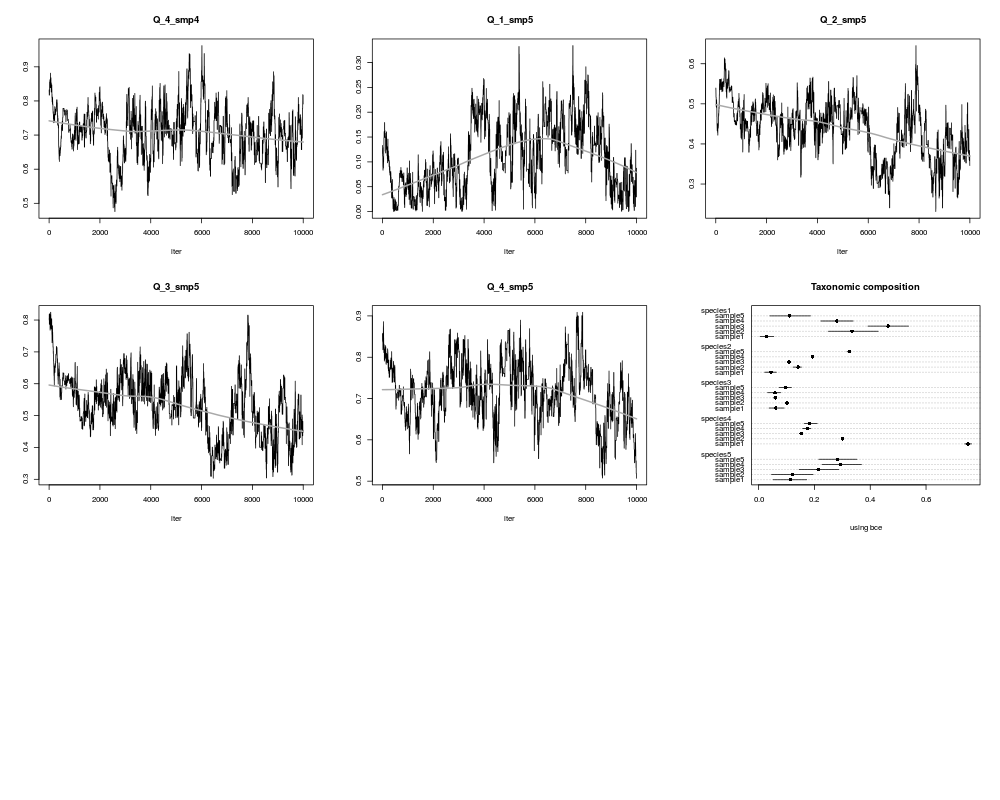
# show results as mean with ranges
print(Sum$meanX)
# plot estimated means and ranges (lbX=lower, ubX=upper bound)
xlim <- range(c(Sum$lbX,Sum$ubX))
# first the mean
dotchart(x=t(Sum$meanX),xlim=xlim,
main="Taxonomic composition",
sub="using bce",pch=16)
# then ranges
nr <- nrow(Sum$meanX)
nc <- ncol(Sum$meanX)
for (i in 1:nr)
{ip <-(nr-i)*(nc+2)+1
cc <- ip : (ip+nc-1)
segments(t(Sum$lbX[i,]),cc,t(Sum$ubX[i,]),cc)
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(BCE)
Loading required package: FME
Loading required package: deSolve
Attaching package: 'deSolve'
The following object is masked from 'package:graphics':
matplot
Loading required package: rootSolve
Loading required package: coda
Loading required package: limSolve
Loading required package: Matrix
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/BCE/bce1.Rd_%03d_medium.png", width=480, height=480)
> ### Name: bce1
> ### Title: Bayesian Composition Estimator
> ### Aliases: bce1 bce
> ### Keywords: models
>
> ### ** Examples
>
> ##====================================
>
> # example using bceInput data
> # !!! should be weighted to correspond better to example of BCE!!!
> A <- t(bceInput$Rat)
> B <- t(bceInput$Dat)
>
>
> result <- bce1(A,B,niter=1000)
number of accepted runs: 0 out of 1000 (0%)
>
> ## the number of accepted runs is zero;
> ## try different starting values
>
> result <- bce1(A,B,niter=1000,initX=matrix(1/ncol(A),ncol(A),ncol(B)))
number of accepted runs: 3 out of 1000 (0.3%)
>
> ## number of accepted runs is still low;
> ## smaller jumps
>
> result <- bce1(A,B,niter=1000,initX=matrix(1/ncol(A),ncol(A),ncol(B)),jmpA=.01,jmpX=.01)
number of accepted runs: 599 out of 1000 (59.9%)
> Sum <-summary(result)
>
> ## did the algorithm converge?
> plot(result$SS,type="l")
> ## no
>
>
> ## more runs, using the output of previous run as input.
> result <- bce1(A,B,niter=1e4,jmpA=.01,jmpX=.01,updatecov=1e3,
+ initX=Sum$lastX,initA=Sum$lastA,
+ jmpCovar=Sum$covar*2.4^2/ncol(result$pars),
+ )
number of accepted runs: 2231 out of 10000 (22.31%)
> Sum <-summary(result)
>
> ## we inspect the output:
> plot(result$SS,type="l")
> plot(result,ask=TRUE)
> ## looks already pretty good; to get a better result, repeat one more
> ## time with a longer run. Uncomment the following paragraph and run.
> ## go get some coffee, this might take a while (~30s).
>
> ## result <- bce1(A,B,niter=1e5,jmpA=.01,jmpX=.01,updatecov=1e3,
> ## outputlength=1e3,burninlength=.35e5,
> ## initX=Sum$lastX,initA=Sum$lastA,
> ## jmpCovar=Sum$covar*2.4^2/ncol(result$pars),
> ## )
> ## Sum <-summary(result)
> ## plot(result$SS,type="l")
> ## plot(result,ask=TRUE)
>
> # show results as mean with ranges
> print(Sum$meanX)
sample1 sample2 sample3 sample4 sample5
species1 0.05961017 0.26185310 0.31148917 0.26198854 0.11634095
species2 0.07009629 0.26325358 0.14119779 0.21569245 0.14203054
species3 0.06221225 0.08024785 0.07674516 0.07383608 0.06040147
species4 0.71671509 0.20937915 0.21846790 0.12876959 0.27164753
species5 0.09136619 0.18526632 0.25209999 0.31971333 0.40957951
>
> # plot estimated means and ranges (lbX=lower, ubX=upper bound)
> xlim <- range(c(Sum$lbX,Sum$ubX))
>
> # first the mean
> dotchart(x=t(Sum$meanX),xlim=xlim,
+ main="Taxonomic composition",
+ sub="using bce",pch=16)
>
> # then ranges
> nr <- nrow(Sum$meanX)
> nc <- ncol(Sum$meanX)
>
> for (i in 1:nr)
+ {ip <-(nr-i)*(nc+2)+1
+ cc <- ip : (ip+nc-1)
+ segments(t(Sum$lbX[i,]),cc,t(Sum$ubX[i,]),cc)
+ }
>
>
>
>
>
>
> dev.off()
null device
1
>
|