Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Highest Density IntervalDescriptionCalculate the highest density interval (HDI) for a given probability mass, see Details. The function is generic, with methods for a range of input objects. Usagehdi(object, credMass = 0.95, ...) ## Default S3 method: hdi(object, credMass = 0.95, ...) ## S3 method for class 'function' hdi(object, credMass = 0.95, tol, ...) ## S3 method for class 'matrix' hdi(object, credMass = 0.95, ...) ## S3 method for class 'data.frame' hdi(object, credMass = 0.95, ...) ## S3 method for class 'density' hdi(object, credMass = 0.95, allowSplit=FALSE, ...) ## S3 method for class 'mcmc.list' hdi(object, credMass = 0.95, ...) Arguments
DetailsThe HDI is the interval which contains the required mass such that all points within the interval have a higher probability density than points outside the interval. When applied to a posterior probability density, it is often known as the Highest Posterior Density (HPD).
In contrast, a symmetric density interval defined by (eg.) the 10% and 90% quantiles may include values with lower probability than those excluded. For a unimodal distribution, the HDI is the narrowest interval containing the specified mass, and the
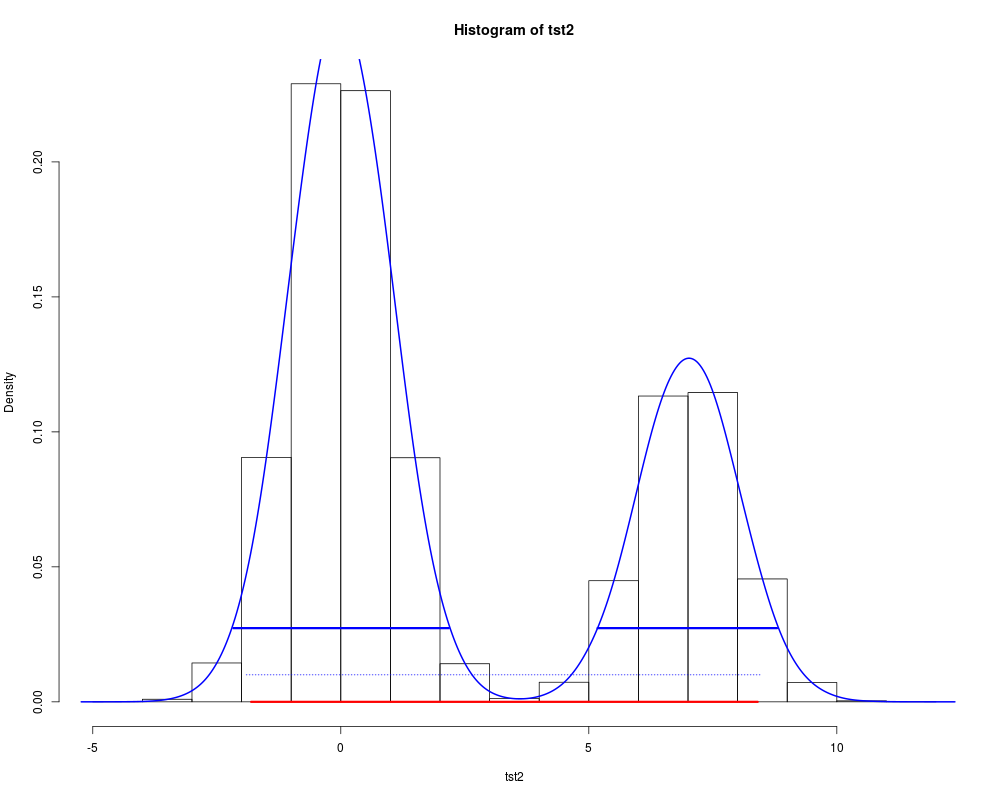
The default method expects a vector representing draws from the target distribution, such as is produced by an MCMC process. Missing values are silently ignored; if the vector has no non-missing values, NAs are returned. The matrix and data frame methods expect an object with vectors of the above type for each parameter in columns. The result is a matrix with parameters in columns, and rows with the upper and lower limits of the HDI. The mcmc.list method expects an object of type None of the above use interpolation: the values returned correspond to specific values in the data object. Results thus depend on the random draws, and will be unstable if few values are provided. For a 95% HDI, 10,000 independent draws are recommended; a smaller number will be adequate for a 80% HDI, many more for a 99% HDI. The function method requires the name for the inverse cumulative density function (ICDF) of the distribution; standard R functions for this have a Valuea vector of length 2 or a 2-row matrix with the lower and upper limits of the HDI, with an attribute "credMass". The Author(s)Mike Meredith. Code for ReferencesKruschke, J. K. 2011. Doing Bayesian data analysis: a tutorial with R and BUGS. Elsevier, Amsterdam, section 3.3.5. Examples# for a vector: tst <- rgamma(1e5, 2.5, 2) hdi(tst) hdi(tst, credMass=0.8) # For comparison, the symmetrical 80% CrI: quantile(tst, c(0.1,0.9)) # Now a data frame: tst <- data.frame(mu = rnorm(1e4, 4, 1), sigma = rlnorm(1e4)) hdi(tst, 0.8) apply(tst, 2, quantile, c(0.1,0.9)) # For a function: hdi(qgamma, 0.8, shape=2.5, rate=2) # and the symmetrical 80% CrI: qgamma(c(0.1, 0.9), 2.5, 2) # A severely bimodal distribution: tst2 <- c(rnorm(1e5), rnorm(5e4, 7)) hist(tst2, freq=FALSE) (hdiMC <- hdi(tst2)) segments(hdiMC[1], 0, hdiMC[2], 0, lwd=3, col='red') # This is a valid 95% CrI, but not a Highest Density Interval dens2 <- density(tst2) lines(dens2, lwd=2, col='blue') (hdiD1 <- hdi(dens2)) # default allowSplit = FALSE; note the warning segments(hdiD1[1], 0.01, hdiD1[2], 0.01, lty=3, col='blue') # This is a valid 95% CrI, but not an HDI (hdiD2 <- hdi(dens2, allowSplit=TRUE)) (ht <- attr(hdiD2, "height")) segments(hdiD2[, 1], ht, hdiD2[, 2], ht, lwd=3, col='blue') # This is the correct 95% HDI. Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(BEST)
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/BEST/hdi.Rd_%03d_medium.png", width=480, height=480)
> ### Name: hdi
> ### Title: Highest Density Interval
> ### Aliases: hdi hdi.default hdi.function hdi.matrix hdi.data.frame
> ### hdi.density hdi.mcmc.list
> ### Keywords: methods htest
>
> ### ** Examples
>
> # for a vector:
> tst <- rgamma(1e5, 2.5, 2)
> hdi(tst)
lower upper
0.09183344 2.82428024
attr(,"credMass")
[1] 0.95
> hdi(tst, credMass=0.8)
lower upper
0.194361 1.903230
attr(,"credMass")
[1] 0.8
> # For comparison, the symmetrical 80% CrI:
> quantile(tst, c(0.1,0.9))
10% 90%
0.403489 2.310669
>
> # Now a data frame:
> tst <- data.frame(mu = rnorm(1e4, 4, 1), sigma = rlnorm(1e4))
> hdi(tst, 0.8)
mu sigma
lower 2.728598 0.05449496
upper 5.238388 2.34642114
attr(,"credMass")
[1] 0.8
> apply(tst, 2, quantile, c(0.1,0.9))
mu sigma
10% 2.728571 0.2833976
90% 5.238360 3.5011073
>
> # For a function:
> hdi(qgamma, 0.8, shape=2.5, rate=2)
lower upper
0.1947158 1.9053925
attr(,"credMass")
[1] 0.8
> # and the symmetrical 80% CrI:
> qgamma(c(0.1, 0.9), 2.5, 2)
[1] 0.402577 2.309089
>
> # A severely bimodal distribution:
> tst2 <- c(rnorm(1e5), rnorm(5e4, 7))
> hist(tst2, freq=FALSE)
> (hdiMC <- hdi(tst2))
lower upper
-1.830860 8.375436
attr(,"credMass")
[1] 0.95
> segments(hdiMC[1], 0, hdiMC[2], 0, lwd=3, col='red')
> # This is a valid 95% CrI, but not a Highest Density Interval
>
> dens2 <- density(tst2)
> lines(dens2, lwd=2, col='blue')
> (hdiD1 <- hdi(dens2)) # default allowSplit = FALSE; note the warning
lower upper
-1.931874 8.437095
attr(,"credMass")
[1] 0.95
attr(,"height")
[1] 0.02747916
Warning message:
In hdi.density(dens2) : The HDI is discontinuous but allowSplit = FALSE;
the result is a valid CrI but not HDI.
> segments(hdiD1[1], 0.01, hdiD1[2], 0.01, lty=3, col='blue')
> # This is a valid 95% CrI, but not an HDI
> (hdiD2 <- hdi(dens2, allowSplit=TRUE))
begin end
[1,] -2.197291 2.190942
[2,] 5.199004 8.808680
attr(,"credMass")
[1] 0.95
attr(,"height")
[1] 0.02747916
> (ht <- attr(hdiD2, "height"))
[1] 0.02747916
> segments(hdiD2[, 1], ht, hdiD2[, 2], ht, lwd=3, col='blue')
> # This is the correct 95% HDI.
>
>
>
>
>
> dev.off()
null device
1
>
|