Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Matching a dictionary of patterns against a referenceDescriptionA set of functions for finding all the occurrences (aka "matches" or "hits") of a set of patterns (aka the dictionary) in a reference sequence or set of reference sequences (aka the subject) The following functions differ in what they return:
This man page shows how to use these functions (aka the See Usage
matchPDict(pdict, subject,
max.mismatch=0, min.mismatch=0, with.indels=FALSE, fixed=TRUE,
algorithm="auto", verbose=FALSE)
countPDict(pdict, subject,
max.mismatch=0, min.mismatch=0, with.indels=FALSE, fixed=TRUE,
algorithm="auto", verbose=FALSE)
whichPDict(pdict, subject,
max.mismatch=0, min.mismatch=0, with.indels=FALSE, fixed=TRUE,
algorithm="auto", verbose=FALSE)
vcountPDict(pdict, subject,
max.mismatch=0, min.mismatch=0, with.indels=FALSE, fixed=TRUE,
algorithm="auto", collapse=FALSE, weight=1L,
verbose=FALSE, ...)
vwhichPDict(pdict, subject,
max.mismatch=0, min.mismatch=0, with.indels=FALSE, fixed=TRUE,
algorithm="auto", verbose=FALSE)
Arguments
DetailsIn this man page, we assume that you know how to preprocess a dictionary
of DNA patterns that can then be used with any of the When using the ValueIf
If
Author(s)H. Pag<c3><83><c2><a8>s ReferencesAho, Alfred V.; Margaret J. Corasick (June 1975). "Efficient string matching: An aid to bibliographic search". Communications of the ACM 18 (6): 333-340. See AlsoPDict-class,
MIndex-class,
matchPDict-inexact,
Examples
## ---------------------------------------------------------------------
## A. A SIMPLE EXAMPLE OF EXACT MATCHING
## ---------------------------------------------------------------------
## Creating the pattern dictionary:
library(drosophila2probe)
dict0 <- DNAStringSet(drosophila2probe)
dict0 # The original dictionary.
length(dict0) # Hundreds of thousands of patterns.
pdict0 <- PDict(dict0) # Store the original dictionary in
# a PDict object (preprocessing).
## Using the pattern dictionary on chromosome 3R:
library(BSgenome.Dmelanogaster.UCSC.dm3)
chr3R <- Dmelanogaster$chr3R # Load chromosome 3R
chr3R
mi0 <- matchPDict(pdict0, chr3R) # Search...
## Looking at the matches:
start_index <- startIndex(mi0) # Get the start index.
length(start_index) # Same as the original dictionary.
start_index[[8220]] # Starts of the 8220th pattern.
end_index <- endIndex(mi0) # Get the end index.
end_index[[8220]] # Ends of the 8220th pattern.
nmatch_per_pat <- elementNROWS(mi0) # Get the number of matches per pattern.
nmatch_per_pat[[8220]]
mi0[[8220]] # Get the matches for the 8220th pattern.
start(mi0[[8220]]) # Equivalent to startIndex(mi0)[[8220]].
sum(nmatch_per_pat) # Total number of matches.
table(nmatch_per_pat)
i0 <- which(nmatch_per_pat == max(nmatch_per_pat))
pdict0[[i0]] # The pattern with most occurrences.
mi0[[i0]] # Its matches as an IRanges object.
Views(chr3R, mi0[[i0]]) # And as an XStringViews object.
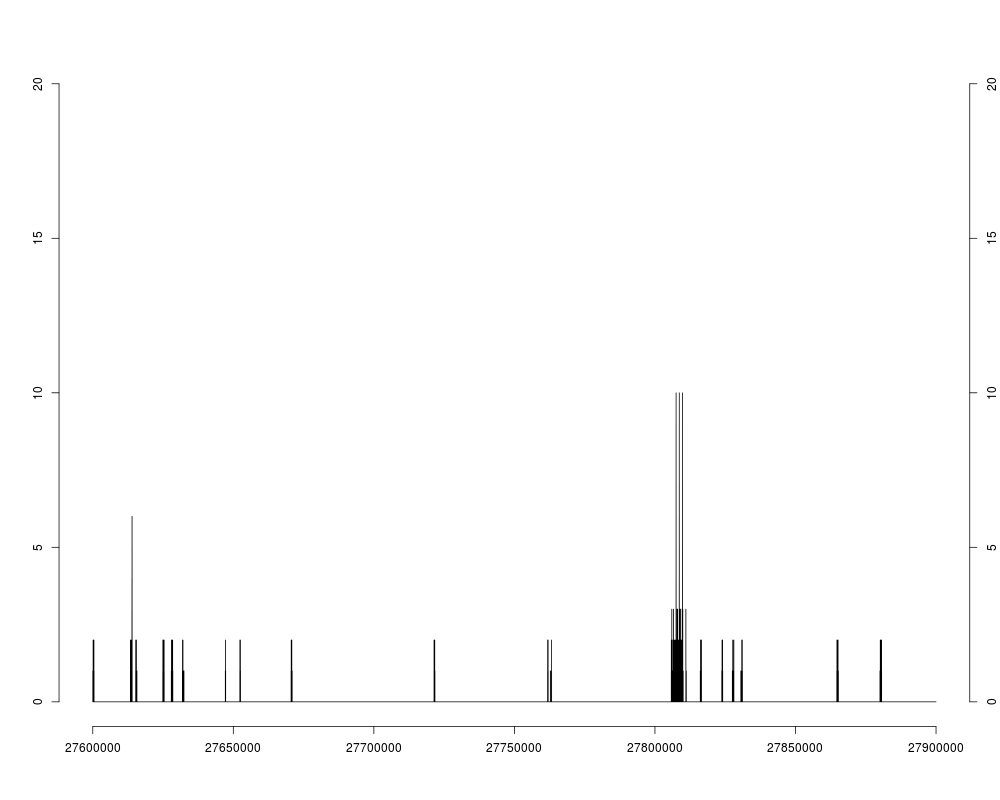
## Get the coverage of the original subject:
cov3R <- as.integer(coverage(mi0, width=length(chr3R)))
max(cov3R)
mean(cov3R)
sum(cov3R != 0) / length(cov3R) # Only 2.44% of chr3R is covered.
if (interactive()) {
plotCoverage <- function(cx, start, end)
{
plot.new()
plot.window(c(start, end), c(0, 20))
axis(1)
axis(2)
axis(4)
lines(start:end, cx[start:end], type="l")
}
plotCoverage(cov3R, 27600000, 27900000)
}
## ---------------------------------------------------------------------
## B. NAMING THE PATTERNS
## ---------------------------------------------------------------------
## The names of the original patterns, if any, are propagated to the
## PDict and MIndex objects:
names(dict0) <- mkAllStrings(letters, 4)[seq_len(length(dict0))]
dict0
dict0[["abcd"]]
pdict0n <- PDict(dict0)
names(pdict0n)[1:30]
pdict0n[["abcd"]]
mi0n <- matchPDict(pdict0n, chr3R)
names(mi0n)[1:30]
mi0n[["abcd"]]
## This is particularly useful when unlisting an MIndex object:
unlist(mi0)[1:10]
unlist(mi0n)[1:10] # keep track of where the matches are coming from
## ---------------------------------------------------------------------
## C. PERFORMANCE
## ---------------------------------------------------------------------
## If getting the number of matches is what matters only (without
## regarding their positions), then countPDict() will be faster,
## especially when there is a high number of matches:
nmatch_per_pat0 <- countPDict(pdict0, chr3R)
stopifnot(identical(nmatch_per_pat0, nmatch_per_pat))
if (interactive()) {
## What's the impact of the dictionary width on performance?
## Below is some code that can be used to figure out (will take a long
## time to run). For different widths of the original dictionary, we
## look at:
## o pptime: preprocessing time (in sec.) i.e. time needed for
## building the PDict object from the truncated input
## sequences;
## o nnodes: nb of nodes in the resulting Aho-Corasick tree;
## o nupatt: nb of unique truncated input sequences;
## o matchtime: time (in sec.) needed to find all the matches;
## o totalcount: total number of matches.
getPDictStats <- function(dict, subject)
{
ans_width <- width(dict[1])
ans_pptime <- system.time(pdict <- PDict(dict))[["elapsed"]]
pptb <- pdict@threeparts@pptb
ans_nnodes <- nnodes(pptb)
ans_nupatt <- sum(!duplicated(pdict))
ans_matchtime <- system.time(
mi0 <- matchPDict(pdict, subject)
)[["elapsed"]]
ans_totalcount <- sum(elementNROWS(mi0))
list(
width=ans_width,
pptime=ans_pptime,
nnodes=ans_nnodes,
nupatt=ans_nupatt,
matchtime=ans_matchtime,
totalcount=ans_totalcount
)
}
stats <- lapply(8:25,
function(width)
getPDictStats(DNAStringSet(dict0, end=width), chr3R))
stats <- data.frame(do.call(rbind, stats))
stats
}
## ---------------------------------------------------------------------
## D. USING A NON-PREPROCESSED DICTIONARY
## ---------------------------------------------------------------------
dict3 <- DNAStringSet(mkAllStrings(DNA_BASES, 3)) # all trinucleotides
dict3
pdict3 <- PDict(dict3)
## The 3 following calls are equivalent (from faster to slower):
res3a <- countPDict(pdict3, chr3R)
res3b <- countPDict(dict3, chr3R)
res3c <- sapply(dict3,
function(pattern) countPattern(pattern, chr3R))
stopifnot(identical(res3a, res3b))
stopifnot(identical(res3a, res3c))
## One reason for using a non-preprocessed dictionary is to get rid of
## all the constraints associated with preprocessing, e.g., when
## preprocessing with PDict(), the input dictionary must be DNA and a
## Trusted Band must be defined (explicitly or implicitly).
## See '?PDict' for more information about these constraints.
## In particular, using a non-preprocessed dictionary can be
## useful for the kind of inexact matching that can't be achieved
## with a PDict object (if performance is not an issue).
## See '?`matchPDict-inexact`' for more information about inexact
## matching.
dictD <- xscat(dict3, "N", reverseComplement(dict3))
## The 2 following calls are equivalent (from faster to slower):
resDa <- matchPDict(dictD, chr3R, fixed=FALSE)
resDb <- sapply(dictD,
function(pattern)
matchPattern(pattern, chr3R, fixed=FALSE))
stopifnot(all(sapply(seq_len(length(dictD)),
function(i)
identical(resDa[[i]], as(resDb[[i]], "IRanges")))))
## A non-preprocessed dictionary can be of any base class i.e. BString,
## RNAString, and AAString, in addition to DNAString:
matchPDict(AAStringSet(c("DARC", "EGH")), AAString("KMFPRNDEGHSTTWTEE"))
## ---------------------------------------------------------------------
## E. vcountPDict()
## ---------------------------------------------------------------------
## Load Fly upstream sequences (i.e. the sequences 2000 bases upstream of
## annotated transcription starts):
dm3_upstream_filepath <- system.file("extdata",
"dm3_upstream2000.fa.gz",
package="Biostrings")
dm3_upstream <- readDNAStringSet(dm3_upstream_filepath)
dm3_upstream
subject <- dm3_upstream[1:100]
mat1 <- vcountPDict(pdict0, subject)
dim(mat1) # length(pdict0) x length(subject)
nhit_per_probe <- rowSums(mat1)
table(nhit_per_probe)
## Without vcountPDict(), 'mat1' could have been computed with:
mat2 <- sapply(unname(subject), function(x) countPDict(pdict0, x))
stopifnot(identical(mat1, mat2))
## but using vcountPDict() is faster (10x or more, depending of the
## average length of the sequences in 'subject').
if (interactive()) {
## This will fail (with message "allocMatrix: too many elements
## specified") because, on most platforms, vectors and matrices in R
## are limited to 2^31 elements:
subject <- dm3_upstream
vcountPDict(pdict0, subject)
length(pdict0) * length(dm3_upstream)
1 * length(pdict0) * length(dm3_upstream) # > 2^31
## But this will work:
nhit_per_seq <- vcountPDict(pdict0, subject, collapse=2)
sum(nhit_per_seq >= 1) # nb of subject sequences with at least 1 hit
table(nhit_per_seq) # max is 74
which.max(nhit_per_seq) # 1133
sum(countPDict(pdict0, subject[[1133]])) # 74
}
## ---------------------------------------------------------------------
## F. RELATIONSHIP BETWEEN vcountPDict(), countPDict() AND
## vcountPattern()
## ---------------------------------------------------------------------
subject <- dm3_upstream
## The 4 following calls are equivalent (from faster to slower):
mat3a <- vcountPDict(pdict3, subject)
mat3b <- vcountPDict(dict3, subject)
mat3c <- sapply(dict3,
function(pattern) vcountPattern(pattern, subject))
mat3d <- sapply(unname(subject),
function(x) countPDict(pdict3, x))
stopifnot(identical(mat3a, mat3b))
stopifnot(identical(mat3a, t(mat3c)))
stopifnot(identical(mat3a, mat3d))
## The 3 following calls are equivalent (from faster to slower):
nhitpp3a <- vcountPDict(pdict3, subject, collapse=1) # rowSums(mat3a)
nhitpp3b <- vcountPDict(dict3, subject, collapse=1)
nhitpp3c <- sapply(dict3,
function(pattern) sum(vcountPattern(pattern, subject)))
stopifnot(identical(nhitpp3a, nhitpp3b))
stopifnot(identical(nhitpp3a, nhitpp3c))
## The 3 following calls are equivalent (from faster to slower):
nhitps3a <- vcountPDict(pdict3, subject, collapse=2) # colSums(mat3a)
nhitps3b <- vcountPDict(dict3, subject, collapse=2)
nhitps3c <- sapply(unname(subject),
function(x) sum(countPDict(pdict3, x)))
stopifnot(identical(nhitps3a, nhitps3b))
stopifnot(identical(nhitps3a, nhitps3c))
## ---------------------------------------------------------------------
## G. vwhichPDict()
## ---------------------------------------------------------------------
subject <- dm3_upstream
## The 4 following calls are equivalent (from faster to slower):
vwp3a <- vwhichPDict(pdict3, subject)
vwp3b <- vwhichPDict(dict3, subject)
vwp3c <- lapply(seq_len(ncol(mat3a)), function(j) which(mat3a[ , j] != 0L))
vwp3d <- lapply(unname(subject), function(x) whichPDict(pdict3, x))
stopifnot(identical(vwp3a, vwp3b))
stopifnot(identical(vwp3a, vwp3c))
stopifnot(identical(vwp3a, vwp3d))
table(sapply(vwp3a, length))
which.min(sapply(vwp3a, length))
## Get the trinucleotides not represented in upstream sequence 21823:
dict3[-vwp3a[[21823]]] # 2 trinucleotides
## Sanity check:
tnf <- trinucleotideFrequency(subject[[21823]])
stopifnot(all(names(tnf)[tnf == 0] == dict3[-vwp3a[[21823]]]))
## ---------------------------------------------------------------------
## H. MAPPING PROBE SET IDS BETWEEN CHIPS WITH vwhichPDict()
## ---------------------------------------------------------------------
## Here we show a simple (and very naive) algorithm for mapping probe
## set IDs between the hgu95av2 and hgu133a chips (Affymetrix).
## 2 probe set IDs are considered mapped iff they share at least one
## probe.
## WARNING: This example takes about 10 minutes to run.
if (interactive()) {
library(hgu95av2probe)
library(hgu133aprobe)
probes1 <- DNAStringSet(hgu95av2probe)
probes2 <- DNAStringSet(hgu133aprobe)
pdict2 <- PDict(probes2)
## Get the mapping from probes1 to probes2 (based on exact matching):
map1to2 <- vwhichPDict(pdict2, probes1)
## The following helper function uses the probe level mapping to induce
## the mapping at the probe set IDs level (from hgu95av2 to hgu133a).
## To keep things simple, 2 probe set IDs are considered mapped iff
## each of them contains at least one probe mapped to one probe of
## the other:
mapProbeSetIDs1to2 <- function(psID)
unique(hgu133aprobe$Probe.Set.Name[unlist(
map1to2[hgu95av2probe$Probe.Set.Name == psID]
)])
## Use the helper function to build the complete mapping:
psIDs1 <- unique(hgu95av2probe$Probe.Set.Name)
mapPSIDs1to2 <- lapply(psIDs1, mapProbeSetIDs1to2) # about 3 min.
names(mapPSIDs1to2) <- psIDs1
## Do some basic stats:
table(sapply(mapPSIDs1to2, length))
## [ADVANCED USERS ONLY]
## An alternative that is slightly faster is to put all the probes
## (hgu95av2 + hgu133a) in a single PDict object and then query its
## 'dups0' slot directly. This slot is a Dups object containing the
## mapping between duplicated patterns.
## Note that we can do this only because all the probes have the
## same length (25) and because we are doing exact matching:
probes12 <- DNAStringSet(c(hgu95av2probe$sequence, hgu133aprobe$sequence))
pdict12 <- PDict(probes12)
dups0 <- pdict12@dups0
mapProbeSetIDs1to2alt <- function(psID)
{
ii1 <- unique(togroup(dups0, which(hgu95av2probe$Probe.Set.Name == psID)))
ii2 <- members(dups0, ii1) - length(probes1)
ii2 <- ii2[ii2 >= 1L]
unique(hgu133aprobe$Probe.Set.Name[ii2])
}
mapPSIDs1to2alt <- lapply(psIDs1, mapProbeSetIDs1to2alt) # about 5 min.
names(mapPSIDs1to2alt) <- psIDs1
## 'mapPSIDs1to2alt' and 'mapPSIDs1to2' contain the same mapping:
stopifnot(identical(lapply(mapPSIDs1to2alt, sort),
lapply(mapPSIDs1to2, sort)))
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(Biostrings)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/Biostrings/matchPDict-exact.Rd_%03d_medium.png", width=480, height=480)
> ### Name: matchPDict
> ### Title: Matching a dictionary of patterns against a reference
> ### Aliases: matchPDict-exact matchPDict matchPDict,XString-method
> ### matchPDict,XStringSet-method matchPDict,XStringViews-method
> ### matchPDict,MaskedXString-method countPDict countPDict,XString-method
> ### countPDict,XStringSet-method countPDict,XStringViews-method
> ### countPDict,MaskedXString-method whichPDict whichPDict,XString-method
> ### whichPDict,XStringSet-method whichPDict,XStringViews-method
> ### whichPDict,MaskedXString-method vmatchPDict vmatchPDict,ANY-method
> ### vmatchPDict,XString-method vmatchPDict,MaskedXString-method
> ### vcountPDict vcountPDict,XString-method vcountPDict,XStringSet-method
> ### vcountPDict,XStringViews-method vcountPDict,MaskedXString-method
> ### vwhichPDict vwhichPDict,XString-method vwhichPDict,XStringSet-method
> ### vwhichPDict,XStringViews-method vwhichPDict,MaskedXString-method
> ### extractAllMatches
> ### Keywords: methods
>
> ### ** Examples
>
> ## ---------------------------------------------------------------------
> ## A. A SIMPLE EXAMPLE OF EXACT MATCHING
> ## ---------------------------------------------------------------------
>
> ## Creating the pattern dictionary:
> library(drosophila2probe)
Loading required package: AnnotationDbi
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> dict0 <- DNAStringSet(drosophila2probe)
> dict0 # The original dictionary.
A DNAStringSet instance of length 265400
width seq
[1] 25 CCTGAATCCTGGCAATGTCATCATC
[2] 25 ATCCTGGCAATGTCATCATCAATGG
[3] 25 ATCAGTTGTCAACGGCTAATACGCG
[4] 25 ATCAATGGCGATTGCCGCGTCTGCA
[5] 25 CCGCGTCTGCAATGTGAGGGCCTAA
... ... ...
[265396] 25 TACTACTTGAGCCACAACCATCTGA
[265397] 25 AGGGACTAAAGAGGCCCCATGCTCT
[265398] 25 CATGCTCTGTCTGGTGTCAGCGCTA
[265399] 25 GTCAGCGCTACATGGTCCAGGACAA
[265400] 25 CCAGGACAAGTATGGACTTCCCCAC
> length(dict0) # Hundreds of thousands of patterns.
[1] 265400
> pdict0 <- PDict(dict0) # Store the original dictionary in
> # a PDict object (preprocessing).
>
> ## Using the pattern dictionary on chromosome 3R:
> library(BSgenome.Dmelanogaster.UCSC.dm3)
Loading required package: BSgenome
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: rtracklayer
> chr3R <- Dmelanogaster$chr3R # Load chromosome 3R
> chr3R
27905053-letter "DNAString" instance
seq: GAATTCTCTCTTGTTGTAGTCTCTTGACAAAATGCA...TATGTCGCAGCGTGACTGTTCGCATTCTAGGAATTC
> mi0 <- matchPDict(pdict0, chr3R) # Search...
>
> ## Looking at the matches:
> start_index <- startIndex(mi0) # Get the start index.
> length(start_index) # Same as the original dictionary.
[1] 265400
> start_index[[8220]] # Starts of the 8220th pattern.
[1] 6788511 9806158 23620037
> end_index <- endIndex(mi0) # Get the end index.
> end_index[[8220]] # Ends of the 8220th pattern.
[1] 6788535 9806182 23620061
> nmatch_per_pat <- elementNROWS(mi0) # Get the number of matches per pattern.
> nmatch_per_pat[[8220]]
[1] 3
> mi0[[8220]] # Get the matches for the 8220th pattern.
IRanges object with 3 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 6788511 6788535 25
[2] 9806158 9806182 25
[3] 23620037 23620061 25
> start(mi0[[8220]]) # Equivalent to startIndex(mi0)[[8220]].
[1] 6788511 9806158 23620037
> sum(nmatch_per_pat) # Total number of matches.
[1] 32993
> table(nmatch_per_pat)
nmatch_per_pat
0 1 2 3 4 5 6 7 8 9 10
233623 31208 341 42 92 44 21 3 8 4 13
12
1
> i0 <- which(nmatch_per_pat == max(nmatch_per_pat))
> pdict0[[i0]] # The pattern with most occurrences.
25-letter "DNAString" instance
seq: AAGGAAGTTGCACGCTGCACTGTCG
> mi0[[i0]] # Its matches as an IRanges object.
IRanges object with 12 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 27806218 27806242 25
[2] 27806784 27806808 25
[3] 27807348 27807372 25
[4] 27807630 27807654 25
[5] 27807912 27807936 25
... ... ... ...
[8] 27809042 27809066 25
[9] 27809325 27809349 25
[10] 27809607 27809631 25
[11] 27809889 27809913 25
[12] 27810997 27811021 25
> Views(chr3R, mi0[[i0]]) # And as an XStringViews object.
Views on a 27905053-letter DNAString subject
subject: GAATTCTCTCTTGTTGTAGTCTCTTGACAAAATG...TGTCGCAGCGTGACTGTTCGCATTCTAGGAATTC
views:
start end width
[1] 27806218 27806242 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[2] 27806784 27806808 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[3] 27807348 27807372 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[4] 27807630 27807654 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[5] 27807912 27807936 25 [AAGGAAGTTGCACGCTGCACTGTCG]
... ... ... ... ...
[8] 27809042 27809066 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[9] 27809325 27809349 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[10] 27809607 27809631 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[11] 27809889 27809913 25 [AAGGAAGTTGCACGCTGCACTGTCG]
[12] 27810997 27811021 25 [AAGGAAGTTGCACGCTGCACTGTCG]
>
> ## Get the coverage of the original subject:
> cov3R <- as.integer(coverage(mi0, width=length(chr3R)))
> max(cov3R)
[1] 10
> mean(cov3R)
[1] 0.02955827
> sum(cov3R != 0) / length(cov3R) # Only 2.44% of chr3R is covered.
[1] 0.02441407
> #if (interactive()) {
> plotCoverage <- function(cx, start, end)
+ {
+ plot.new()
+ plot.window(c(start, end), c(0, 20))
+ axis(1)
+ axis(2)
+ axis(4)
+ lines(start:end, cx[start:end], type="l")
+ }
> plotCoverage(cov3R, 27600000, 27900000)
> #}
>
> ## ---------------------------------------------------------------------
> ## B. NAMING THE PATTERNS
> ## ---------------------------------------------------------------------
>
> ## The names of the original patterns, if any, are propagated to the
> ## PDict and MIndex objects:
> names(dict0) <- mkAllStrings(letters, 4)[seq_len(length(dict0))]
> dict0
A DNAStringSet instance of length 265400
width seq names
[1] 25 CCTGAATCCTGGCAATGTCATCATC aaaa
[2] 25 ATCCTGGCAATGTCATCATCAATGG aaab
[3] 25 ATCAGTTGTCAACGGCTAATACGCG aaac
[4] 25 ATCAATGGCGATTGCCGCGTCTGCA aaad
[5] 25 CCGCGTCTGCAATGTGAGGGCCTAA aaae
... ... ...
[265396] 25 TACTACTTGAGCCACAACCATCTGA pcpn
[265397] 25 AGGGACTAAAGAGGCCCCATGCTCT pcpo
[265398] 25 CATGCTCTGTCTGGTGTCAGCGCTA pcpp
[265399] 25 GTCAGCGCTACATGGTCCAGGACAA pcpq
[265400] 25 CCAGGACAAGTATGGACTTCCCCAC pcpr
> dict0[["abcd"]]
25-letter "DNAString" instance
seq: CCTCTCAGAGAAACCCACACAAAAA
> pdict0n <- PDict(dict0)
> names(pdict0n)[1:30]
[1] "aaaa" "aaab" "aaac" "aaad" "aaae" "aaaf" "aaag" "aaah" "aaai" "aaaj"
[11] "aaak" "aaal" "aaam" "aaan" "aaao" "aaap" "aaaq" "aaar" "aaas" "aaat"
[21] "aaau" "aaav" "aaaw" "aaax" "aaay" "aaaz" "aaba" "aabb" "aabc" "aabd"
> pdict0n[["abcd"]]
25-letter "DNAString" instance
seq: CCTCTCAGAGAAACCCACACAAAAA
> mi0n <- matchPDict(pdict0n, chr3R)
> names(mi0n)[1:30]
[1] "aaaa" "aaab" "aaac" "aaad" "aaae" "aaaf" "aaag" "aaah" "aaai" "aaaj"
[11] "aaak" "aaal" "aaam" "aaan" "aaao" "aaap" "aaaq" "aaar" "aaas" "aaat"
[21] "aaau" "aaav" "aaaw" "aaax" "aaay" "aaaz" "aaba" "aabb" "aabc" "aabd"
> mi0n[["abcd"]]
IRanges object with 0 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
>
> ## This is particularly useful when unlisting an MIndex object:
> unlist(mi0)[1:10]
IRanges object with 10 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 16069898 16069922 25
[2] 16069961 16069985 25
[3] 16070066 16070090 25
[4] 16070081 16070105 25
[5] 16070100 16070124 25
[6] 16070112 16070136 25
[7] 16070136 16070160 25
[8] 16070159 16070183 25
[9] 16070174 16070198 25
[10] 16070192 16070216 25
> unlist(mi0n)[1:10] # keep track of where the matches are coming from
IRanges object with 10 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
aadu 16069898 16069922 25
aadv 16069961 16069985 25
aadw 16070066 16070090 25
aadx 16070081 16070105 25
aady 16070100 16070124 25
aadz 16070112 16070136 25
aaea 16070136 16070160 25
aaeb 16070159 16070183 25
aaec 16070174 16070198 25
aaed 16070192 16070216 25
>
> ## ---------------------------------------------------------------------
> ## C. PERFORMANCE
> ## ---------------------------------------------------------------------
>
> ## If getting the number of matches is what matters only (without
> ## regarding their positions), then countPDict() will be faster,
> ## especially when there is a high number of matches:
>
> nmatch_per_pat0 <- countPDict(pdict0, chr3R)
> stopifnot(identical(nmatch_per_pat0, nmatch_per_pat))
>
> #if (interactive()) {
> ## What's the impact of the dictionary width on performance?
> ## Below is some code that can be used to figure out (will take a long
> ## time to run). For different widths of the original dictionary, we
> ## look at:
> ## o pptime: preprocessing time (in sec.) i.e. time needed for
> ## building the PDict object from the truncated input
> ## sequences;
> ## o nnodes: nb of nodes in the resulting Aho-Corasick tree;
> ## o nupatt: nb of unique truncated input sequences;
> ## o matchtime: time (in sec.) needed to find all the matches;
> ## o totalcount: total number of matches.
> getPDictStats <- function(dict, subject)
+ {
+ ans_width <- width(dict[1])
+ ans_pptime <- system.time(pdict <- PDict(dict))[["elapsed"]]
+ pptb <- pdict@threeparts@pptb
+ ans_nnodes <- nnodes(pptb)
+ ans_nupatt <- sum(!duplicated(pdict))
+ ans_matchtime <- system.time(
+ mi0 <- matchPDict(pdict, subject)
+ )[["elapsed"]]
+ ans_totalcount <- sum(elementNROWS(mi0))
+ list(
+ width=ans_width,
+ pptime=ans_pptime,
+ nnodes=ans_nnodes,
+ nupatt=ans_nupatt,
+ matchtime=ans_matchtime,
+ totalcount=ans_totalcount
+ )
+ }
> stats <- lapply(8:25,
+ function(width)
+ getPDictStats(DNAStringSet(dict0, end=width), chr3R))
> stats <- data.frame(do.call(rbind, stats))
> stats
width pptime nnodes nupatt matchtime totalcount
1 8 0.36 76736 55394 10.436 137694153
2 9 0.25 211661 134925 6.748 36016949
3 10 0.241 421855 210194 6.597 9486993
4 11 0.216 668678 246823 5.694 2554921
5 12 0.191 927909 259231 5.437 719613
6 13 0.25 1190999 263090 4.92 222616
7 14 0.226 1455249 264250 5.174 85065
8 15 0.246 1719897 264648 5.082 47455
9 16 0.242 1984806 264909 5.07 36521
10 17 0.258 2249717 264911 5.132 34391
11 18 0.263 2514630 264913 5.104 33756
12 19 0.299 2779544 264914 5.028 33499
13 20 0.298 3044460 264916 5.201 33354
14 21 0.288 3309377 264917 4.773 33261
15 22 0.291 3574294 264917 4.817 33190
16 23 0.351 3839212 264918 5.053 33123
17 24 0.231 4104130 264918 4.827 33057
18 25 0.211 4369049 264919 4.69 32993
> #}
>
> ## ---------------------------------------------------------------------
> ## D. USING A NON-PREPROCESSED DICTIONARY
> ## ---------------------------------------------------------------------
>
> dict3 <- DNAStringSet(mkAllStrings(DNA_BASES, 3)) # all trinucleotides
> dict3
A DNAStringSet instance of length 64
width seq
[1] 3 AAA
[2] 3 AAC
[3] 3 AAG
[4] 3 AAT
[5] 3 ACA
... ... ...
[60] 3 TGT
[61] 3 TTA
[62] 3 TTC
[63] 3 TTG
[64] 3 TTT
> pdict3 <- PDict(dict3)
>
> ## The 3 following calls are equivalent (from faster to slower):
> res3a <- countPDict(pdict3, chr3R)
> res3b <- countPDict(dict3, chr3R)
> res3c <- sapply(dict3,
+ function(pattern) countPattern(pattern, chr3R))
> stopifnot(identical(res3a, res3b))
> stopifnot(identical(res3a, res3c))
>
> ## One reason for using a non-preprocessed dictionary is to get rid of
> ## all the constraints associated with preprocessing, e.g., when
> ## preprocessing with PDict(), the input dictionary must be DNA and a
> ## Trusted Band must be defined (explicitly or implicitly).
> ## See '?PDict' for more information about these constraints.
> ## In particular, using a non-preprocessed dictionary can be
> ## useful for the kind of inexact matching that can't be achieved
> ## with a PDict object (if performance is not an issue).
> ## See '?`matchPDict-inexact`' for more information about inexact
> ## matching.
>
> dictD <- xscat(dict3, "N", reverseComplement(dict3))
>
> ## The 2 following calls are equivalent (from faster to slower):
> resDa <- matchPDict(dictD, chr3R, fixed=FALSE)
> resDb <- sapply(dictD,
+ function(pattern)
+ matchPattern(pattern, chr3R, fixed=FALSE))
> stopifnot(all(sapply(seq_len(length(dictD)),
+ function(i)
+ identical(resDa[[i]], as(resDb[[i]], "IRanges")))))
>
> ## A non-preprocessed dictionary can be of any base class i.e. BString,
> ## RNAString, and AAString, in addition to DNAString:
> matchPDict(AAStringSet(c("DARC", "EGH")), AAString("KMFPRNDEGHSTTWTEE"))
MIndex object of length 2
[[1]]
IRanges object with 0 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[[2]]
IRanges object with 1 range and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 8 10 3
>
> ## ---------------------------------------------------------------------
> ## E. vcountPDict()
> ## ---------------------------------------------------------------------
>
> ## Load Fly upstream sequences (i.e. the sequences 2000 bases upstream of
> ## annotated transcription starts):
> dm3_upstream_filepath <- system.file("extdata",
+ "dm3_upstream2000.fa.gz",
+ package="Biostrings")
> dm3_upstream <- readDNAStringSet(dm3_upstream_filepath)
> dm3_upstream
A DNAStringSet instance of length 26454
width seq names
[1] 2000 GTTGGTGGCCCACCAGTGCCA...CAAGTTTACCGGTTGCACGGT NM_078863_up_2000...
[2] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201794_up_2...
[3] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201795_up_2...
[4] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201796_up_2...
[5] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201797_up_2...
... ... ...
[26450] 2000 ATTTACAAGACTAATAAAGAT...AAAATTAAATTTCAATAAAAC NM_001111010_up_2...
[26451] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001015258_up_2...
[26452] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001110997_up_2...
[26453] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001276245_up_2...
[26454] 2000 CGTATGTATTAGTTAACTCTG...TCAAAGTGTAAGAACAAATTG NM_001015497_up_2...
>
> subject <- dm3_upstream[1:100]
> mat1 <- vcountPDict(pdict0, subject)
> dim(mat1) # length(pdict0) x length(subject)
[1] 265400 100
> nhit_per_probe <- rowSums(mat1)
> table(nhit_per_probe)
nhit_per_probe
0 1 2 3 8 9
265217 82 59 14 14 14
>
> ## Without vcountPDict(), 'mat1' could have been computed with:
> mat2 <- sapply(unname(subject), function(x) countPDict(pdict0, x))
> stopifnot(identical(mat1, mat2))
> ## but using vcountPDict() is faster (10x or more, depending of the
> ## average length of the sequences in 'subject').
>
> #if (interactive()) {
> ## This will fail (with message "allocMatrix: too many elements
> ## specified") because, on most platforms, vectors and matrices in R
> ## are limited to 2^31 elements:
> subject <- dm3_upstream
> vcountPDict(pdict0, subject)
Error: cannot allocate vector of size 26.2 Gb
Execution halted
|