Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
DBChIPDescriptionDetecting differential binding of transcription factors with ChIP-seq Usage
DBChIP(binding.site.list, chip.data.list, conds, input.data.list = NULL,
data.type = c("MCS", "AlignedRead", "BED"), frag.len = 200, chr.vec = NULL,
chr.exclusion = NULL, chr.len.vec = NULL, subtract.input = FALSE, norm.factor.vec = NULL,
in.distance = 100, out.distance = 250, window.size = 250,
dispersion=NULL, common.disp=TRUE, prior.n=10,
two.sample.method="composite.null", allowable.FC=1.5, collapsed.quant=0.5)
Arguments
DetailsThe ChIP and control data should be properly filtered before the analysis to avoid artifacts.
For example, reads mapping to mitochondrial DNA, or Y chromosome for female samples will need to be filtered. Filtering of chromosomes can be achieved through specification of User can include or exclude sex chromosomes in the computation, depending on whether protein-DNA bindings on sex chromosomes are of research interest. Biological replicates of a ChIP sample should be kept separate so that dispersion can be properly estimated. On the other hand, replicates of a control/input sample should be merged because the purpose of the control samples is to estimate the background for testing and plotting. One exception would be when a control replicate is paired with a ChIP replicate, for example, they are coming from the same batch, a portion of which is used for IP and the other portion is used for control. In such case, the control replicate can be kept separate with the same name of the matching ChIP replicate. data.type
Users are recommended to study the histogram of the $p$-values for model checking. More specifically, the $p$-values between 0.5 and 1 should be roughly uniform. When many replicates are available, users can also randomly split biological replicates of the same condition and perform comparisons through DBChIP using the estimated dispersion parameter to check whether the $p$-values look uniform. ValueA list with following components:
Author(s)Kun Liang, kliang@stat.wisc.edu ReferencesLiang, K and Keles, S (2012). Detecting differential binding of transcription factors with ChIP-seq. Bioinformatics, 28, 121-122. See Also
Examples
data("PHA4")
dat <- DBChIP(binding.site.list, chip.data.list=chip.data.list, input.data.list=input.data.list, conds=conds, data.type="MCS")
rept <- report.peak(dat)
rept
#pdf("Diff.Binding.pdf")
plotPeak(rept, dat)
#dev.off()
## experienced users can proceed in a step by step fashion such that if program
## needs to be run for a different setting, intermediate results can be saved and reused.
data("PHA4")
conds <- factor(c("emb","emb","L1", "L1"), levels=c("emb", "L1"))
bs.list <- read.binding.site.list(binding.site.list)
## compute consensus site
consensus.site <- site.merge(bs.list, in.distance=100, out.distance=250)
dat <- load.data(chip.data.list=chip.data.list, conds=conds, consensus.site=consensus.site, input.data.list=input.data.list, data.type="MCS")
## count ChIP reads around each binding site
dat <- get.site.count(dat, window.size=250)
## test for differential binding
dat <- test.diff.binding(dat)
# report test result and plot the coverage profiles
rept <- report.peak(dat)
rept
plotPeak(rept, dat)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(DBChIP)
Loading required package: edgeR
Loading required package: limma
Loading required package: DESeq
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following object is masked from 'package:limma':
plotMA
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: locfit
locfit 1.5-9.1 2013-03-22
Loading required package: lattice
Welcome to 'DESeq'. For improved performance, usability and
functionality, please consider migrating to 'DESeq2'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/DBChIP/DBChIP.Rd_%03d_medium.png", width=480, height=480)
> ### Name: DBChIP
> ### Title: DBChIP
> ### Aliases: DBChIP
>
> ### ** Examples
>
> data("PHA4")
> dat <- DBChIP(binding.site.list, chip.data.list=chip.data.list, input.data.list=input.data.list, conds=conds, data.type="MCS")
merging sites from different conditions to consensus sites.done
reading data...done
computing normalization factor between ChIP and control samples.done
count ChIP reads around each binding site.done
Common dispersion: 0.05915362
> rept <- report.peak(dat)
> rept
chr pos nsig origin ori.pos FC.L1 pval FDR
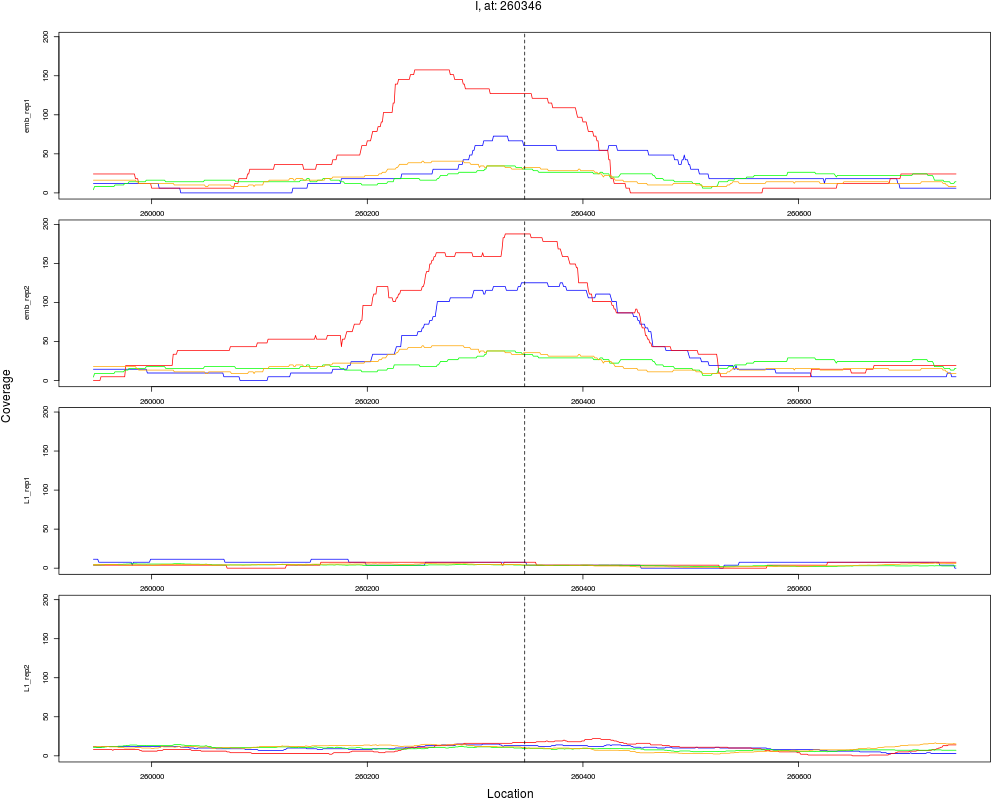
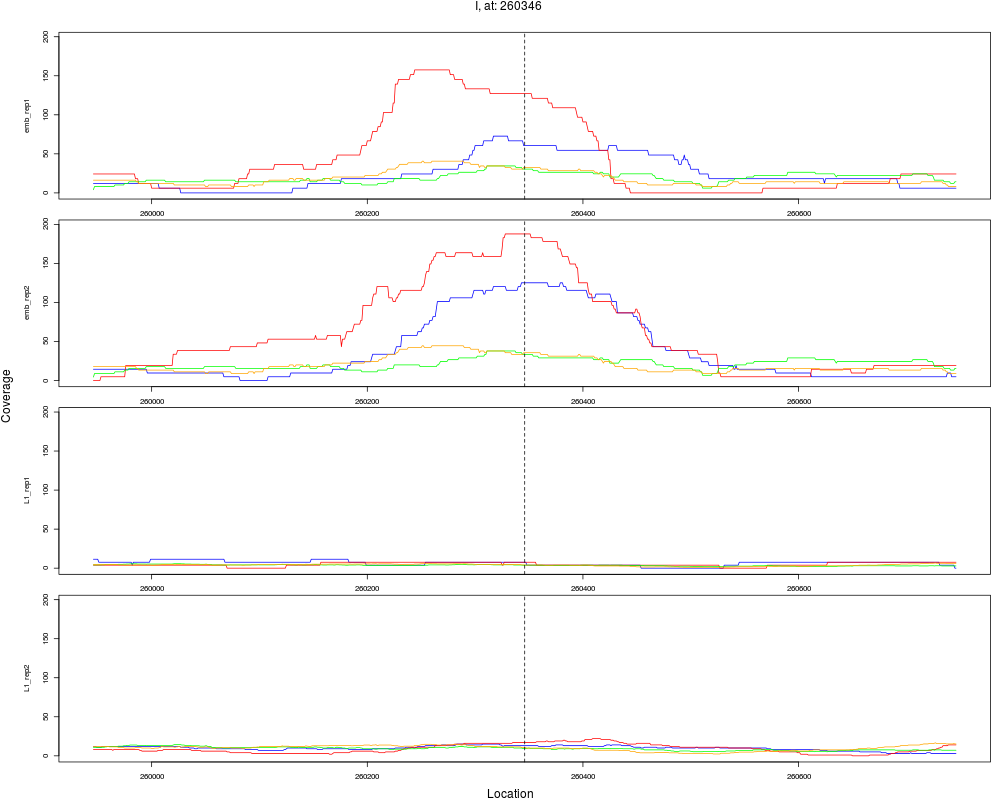
1 I 260346 1 emb 260346 0.1129558 1.057645e-11 5.076698e-10
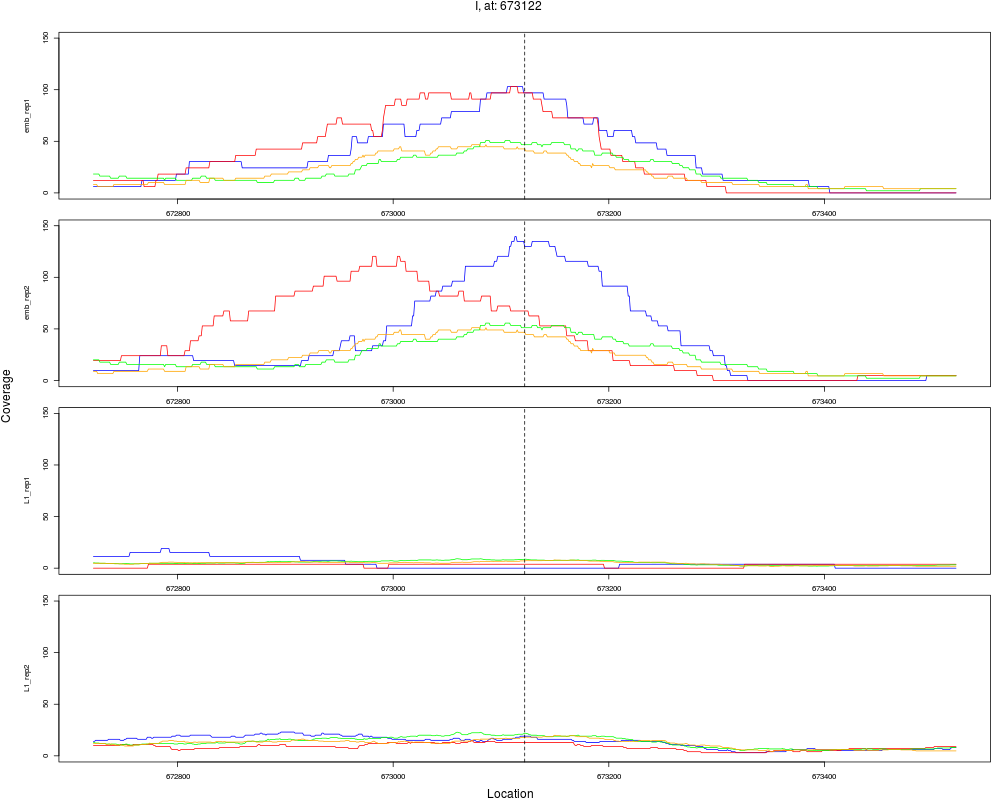
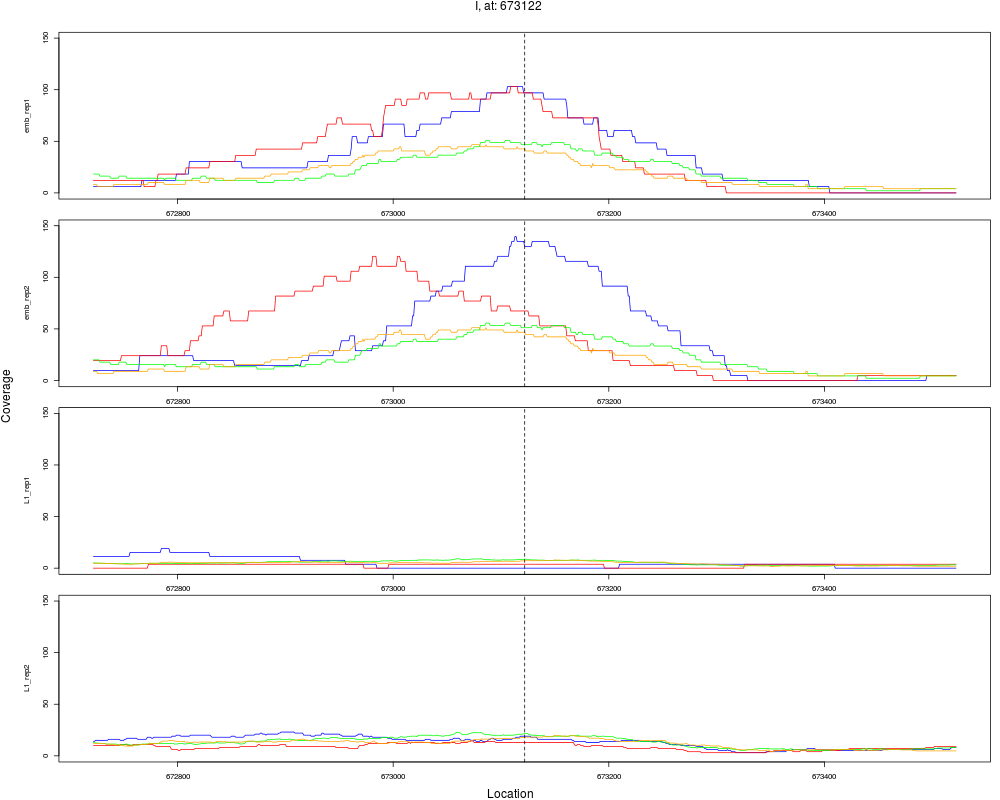
2 I 673122 1 emb 673122 0.1168590 1.798443e-10 4.316264e-09
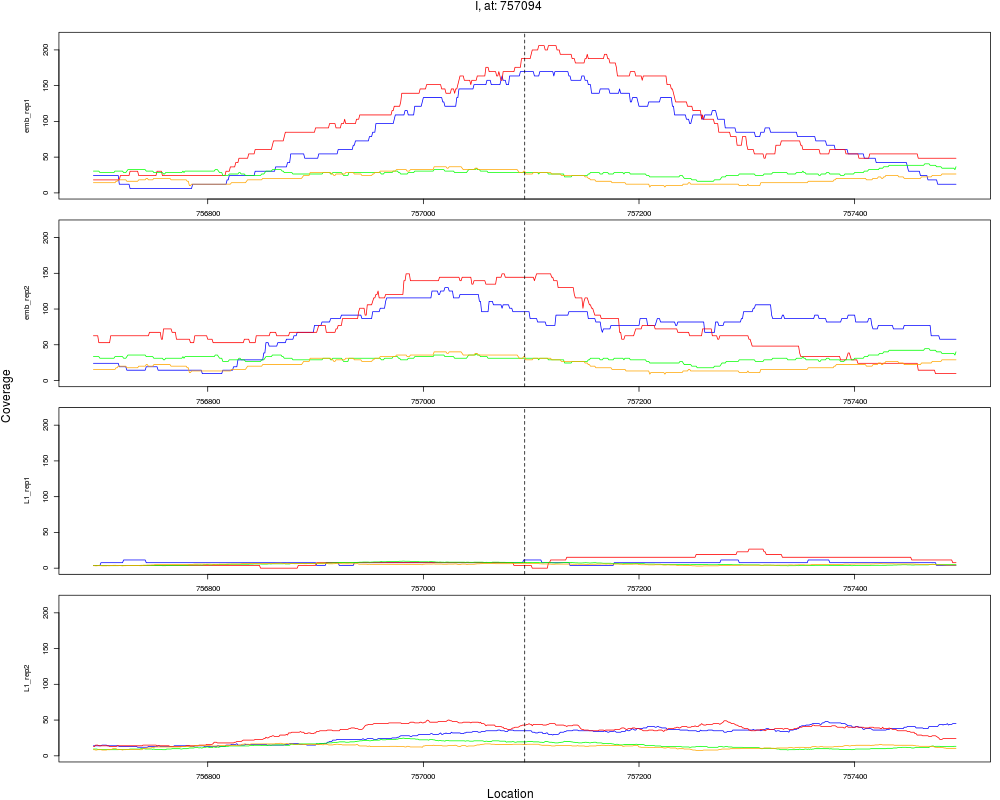
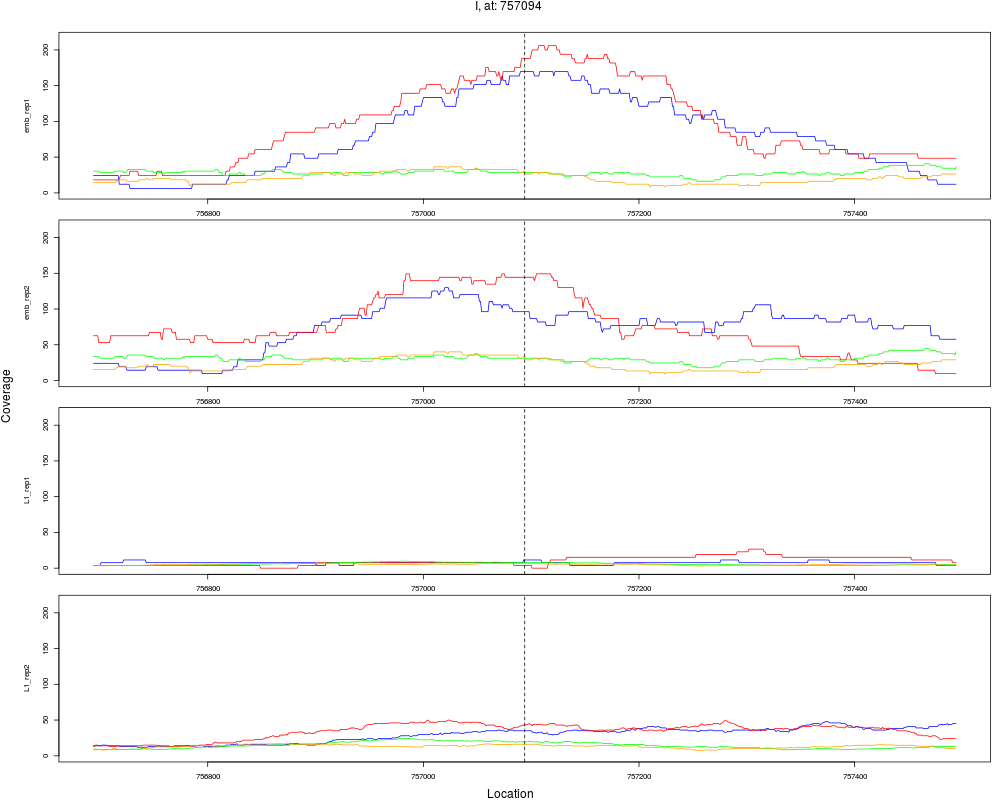
3 I 757094 1 emb 757094 0.1717831 4.012809e-09 6.420495e-08
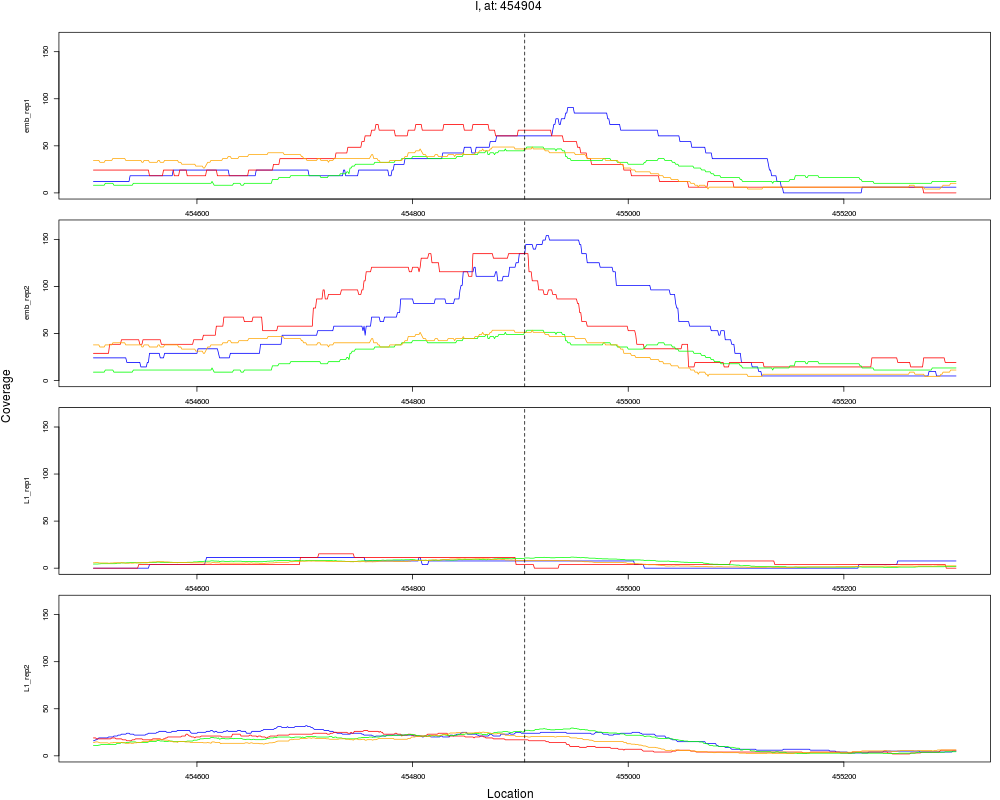
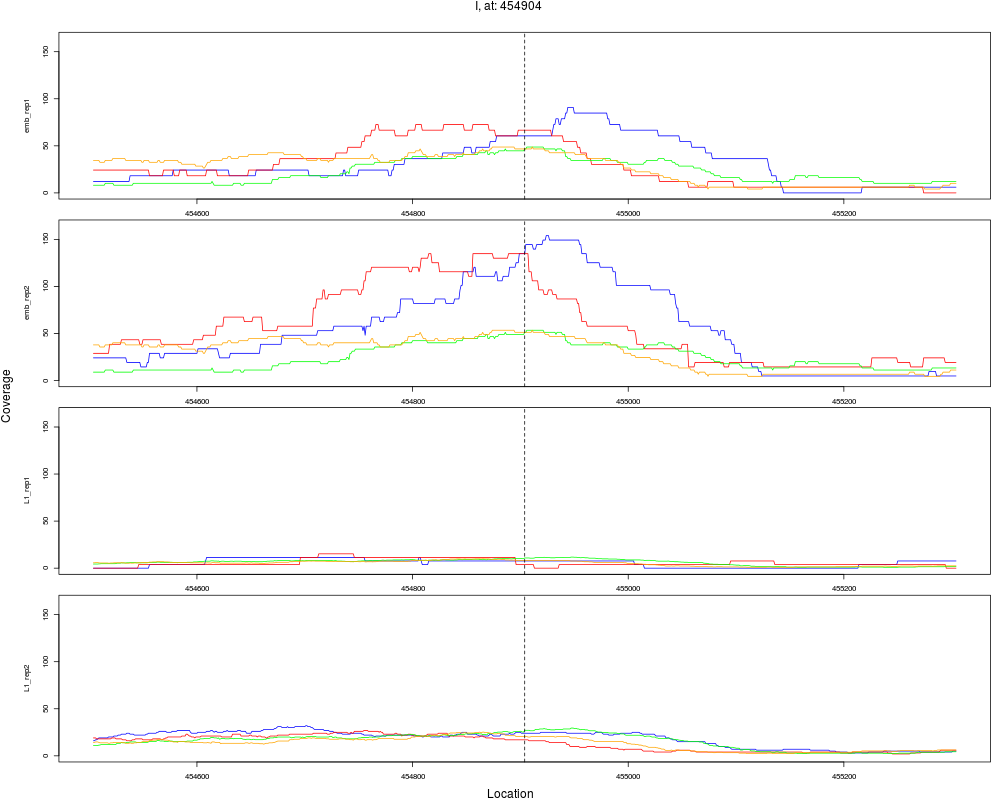
4 I 454904 1 emb 454904 0.1632294 1.349896e-08 1.619875e-07
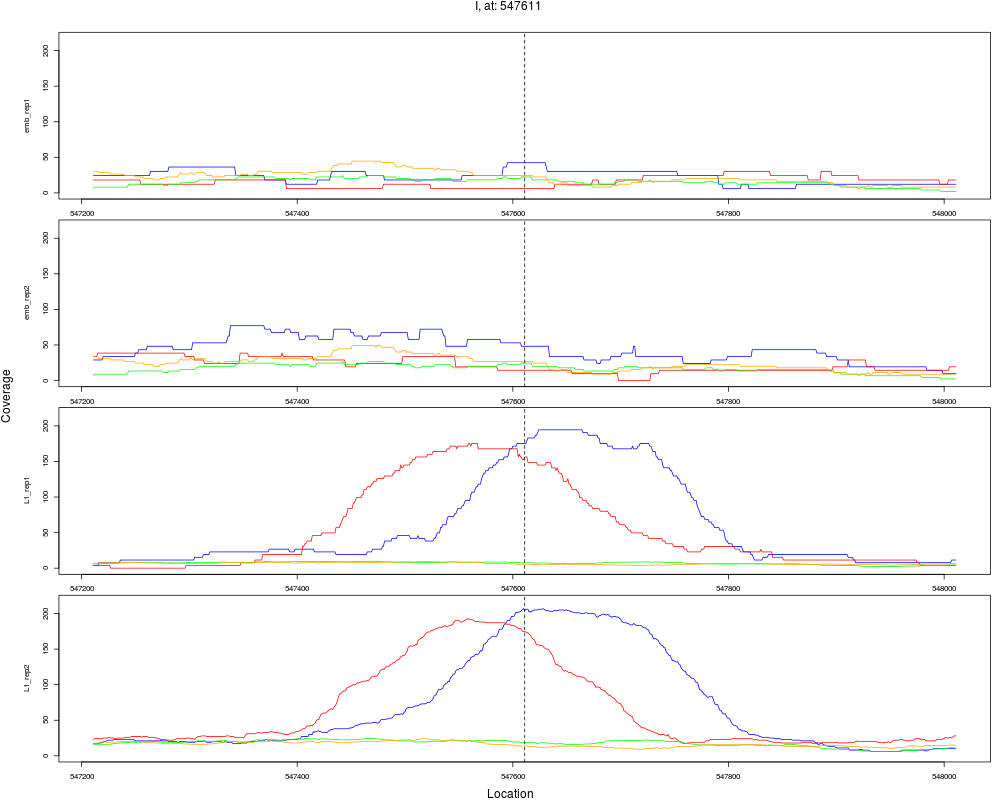
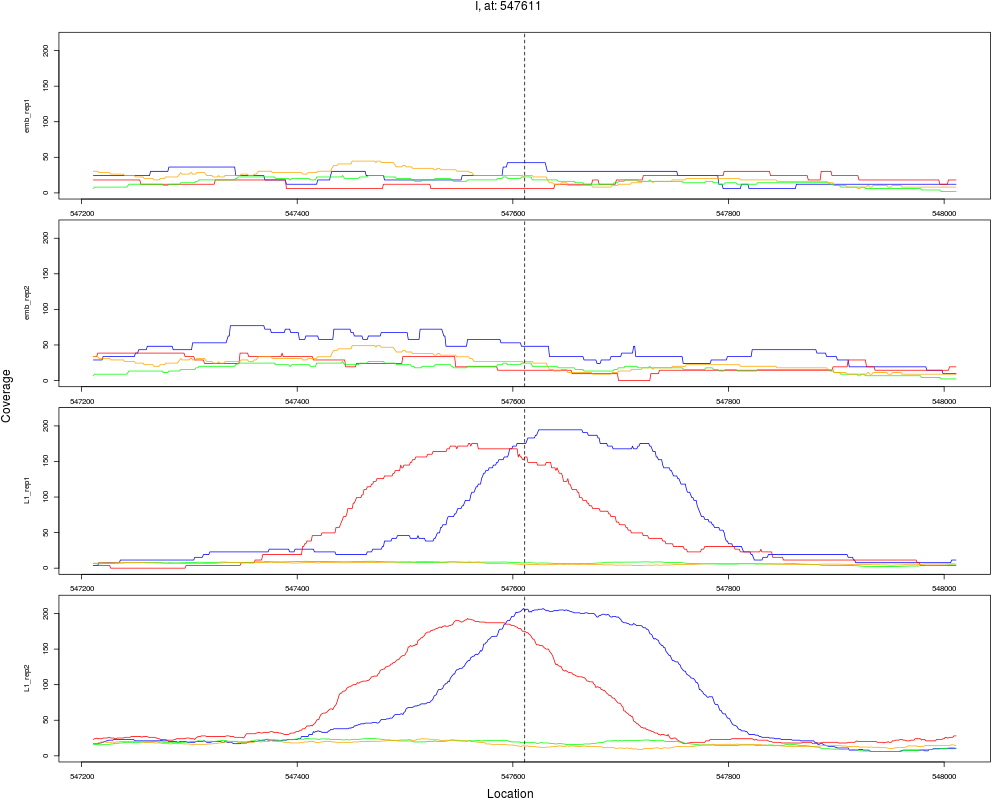
5 I 547611 1 L1 547611 5.6827777 2.104486e-08 2.020307e-07
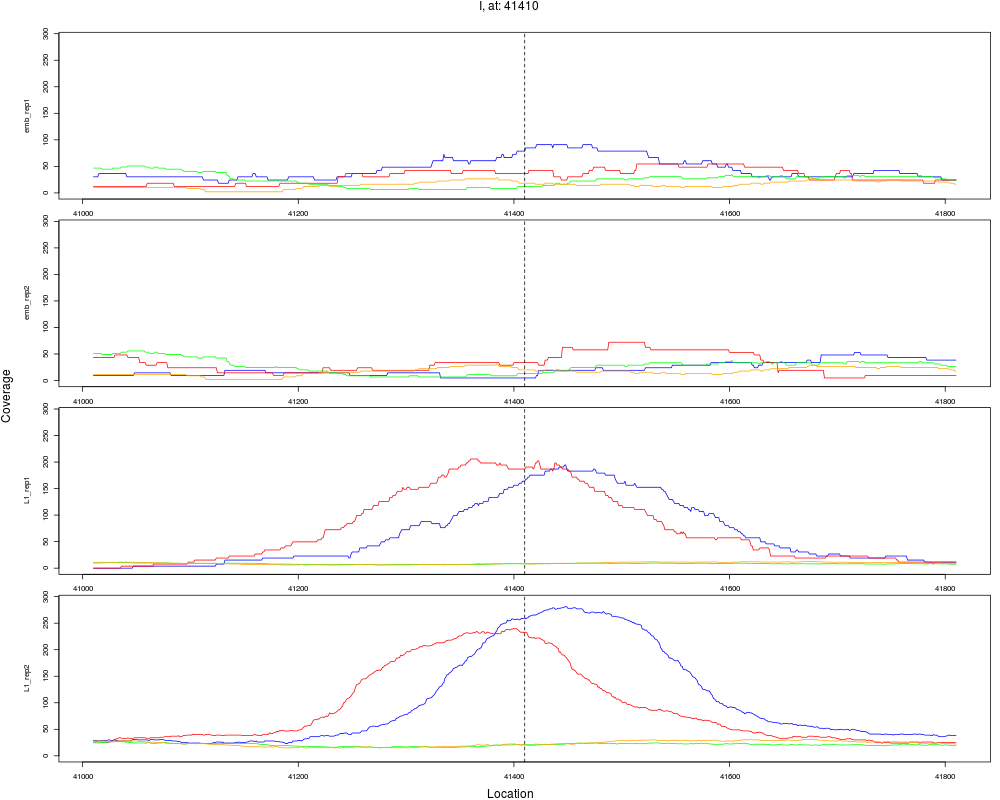
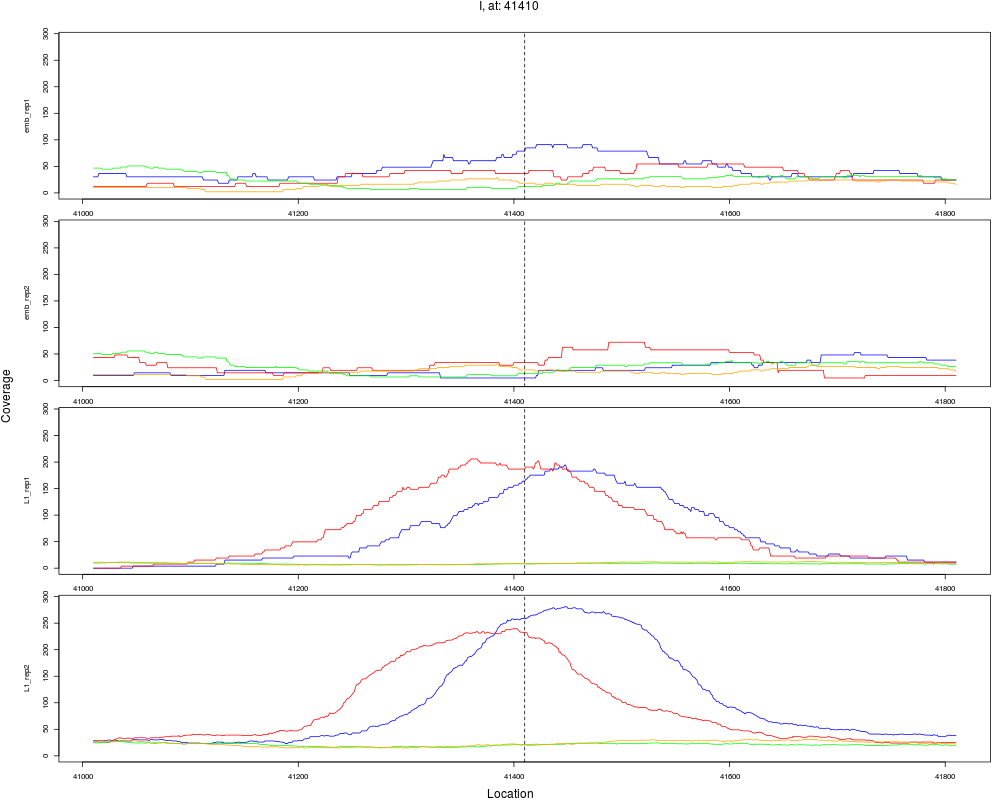
6 I 41410 1 L1 41410 4.2984821 4.548449e-07 3.638759e-06
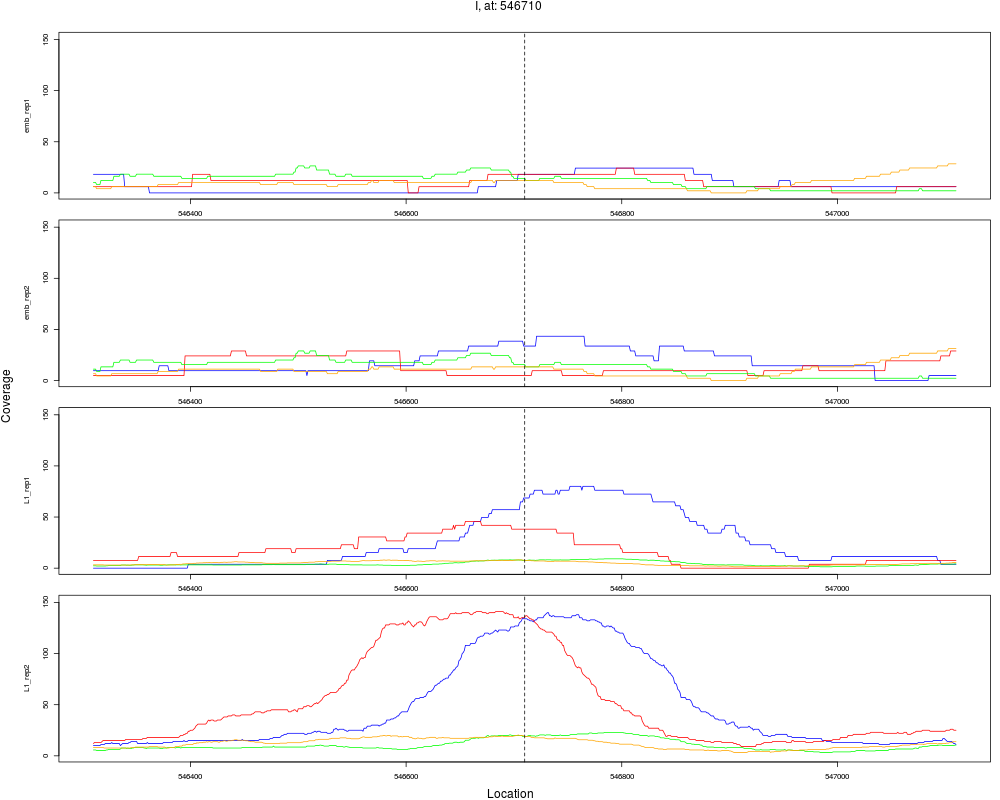
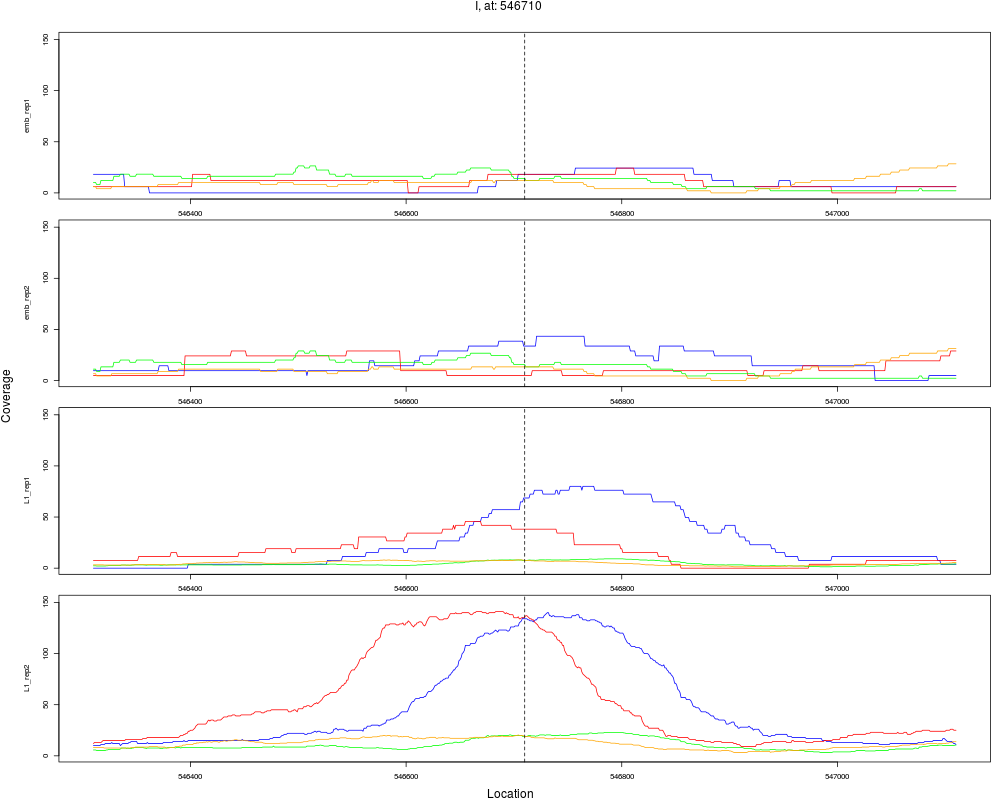
7 I 546710 1 L1 546710 4.3025538 7.621600e-06 5.226240e-05
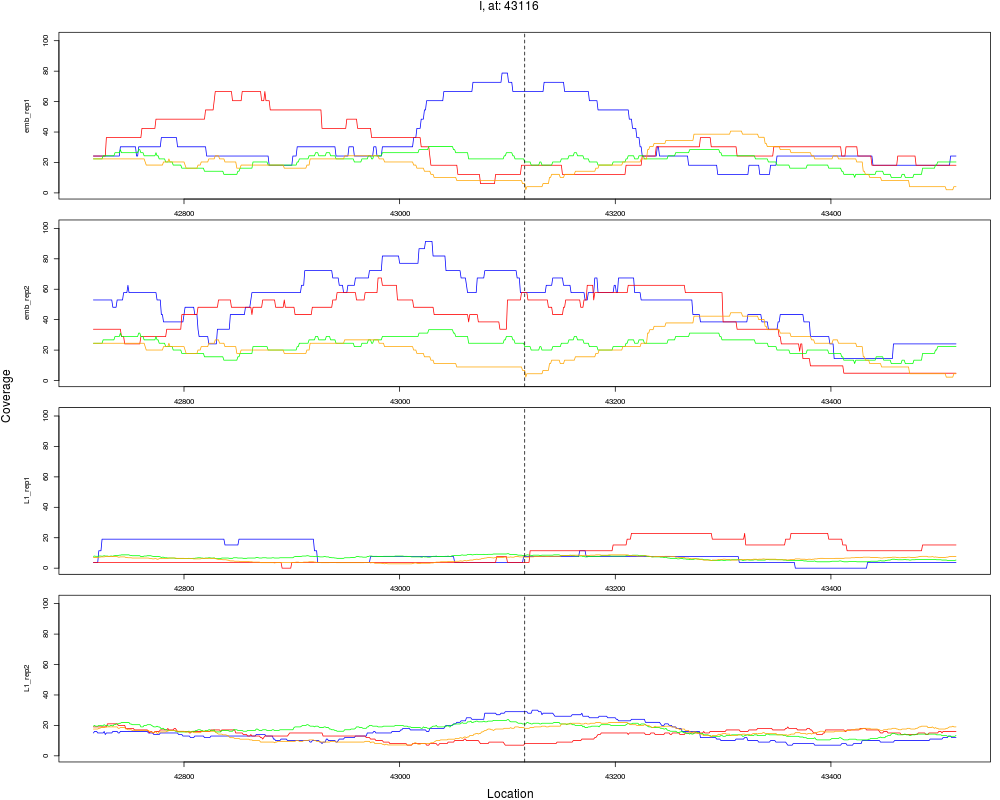
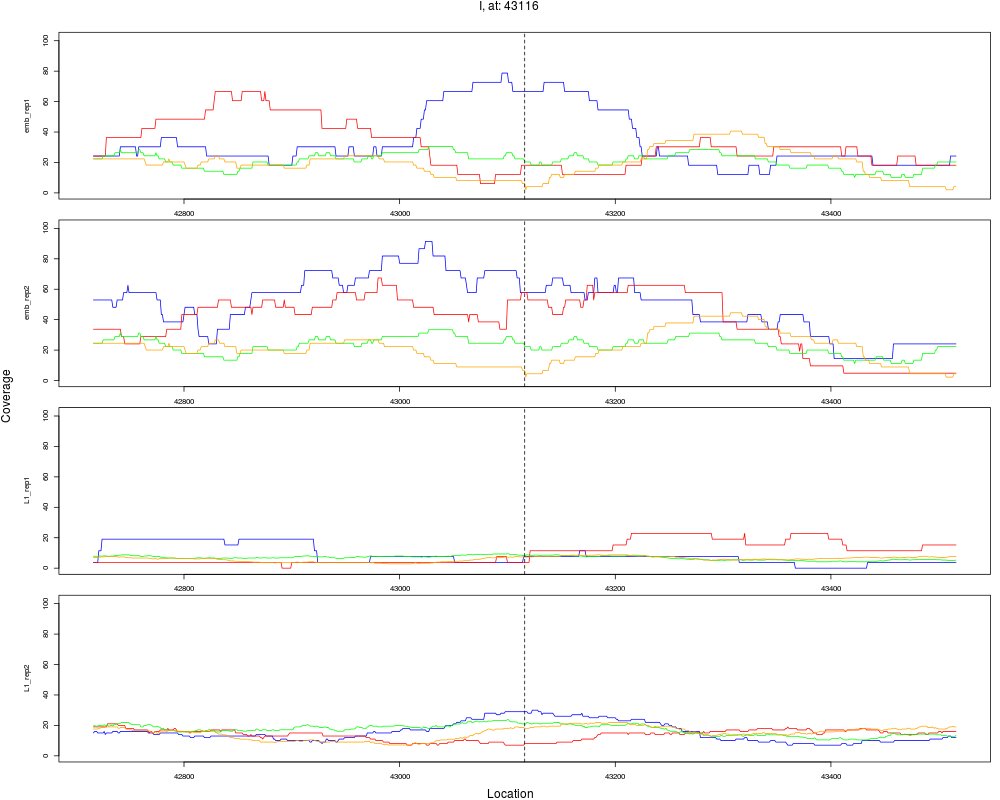
8 I 43116 1 emb 43116 0.2581921 6.466525e-05 3.879915e-04
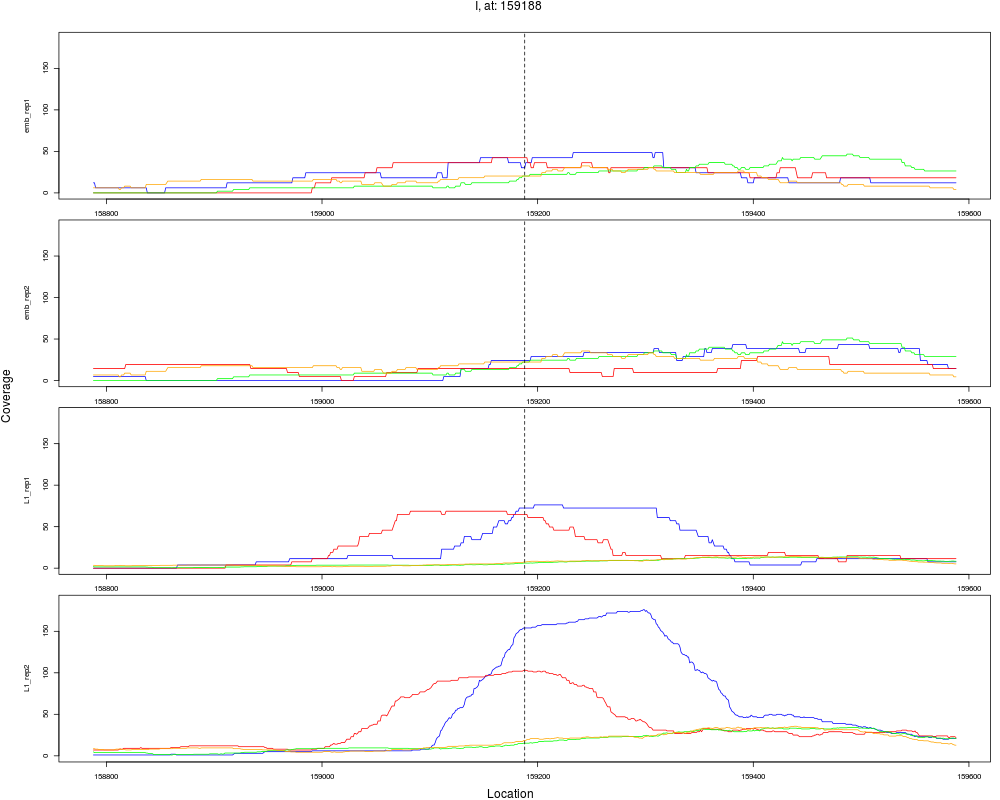
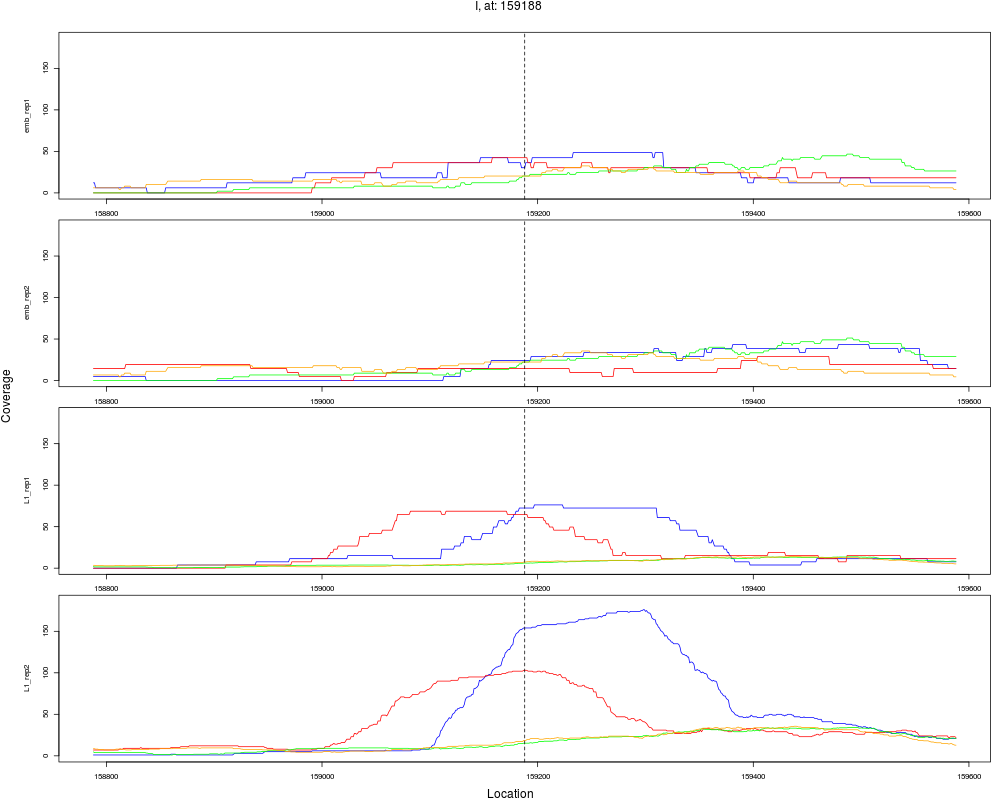
9 I 159188 1 L1 159188 2.9888809 3.449015e-04 1.839475e-03
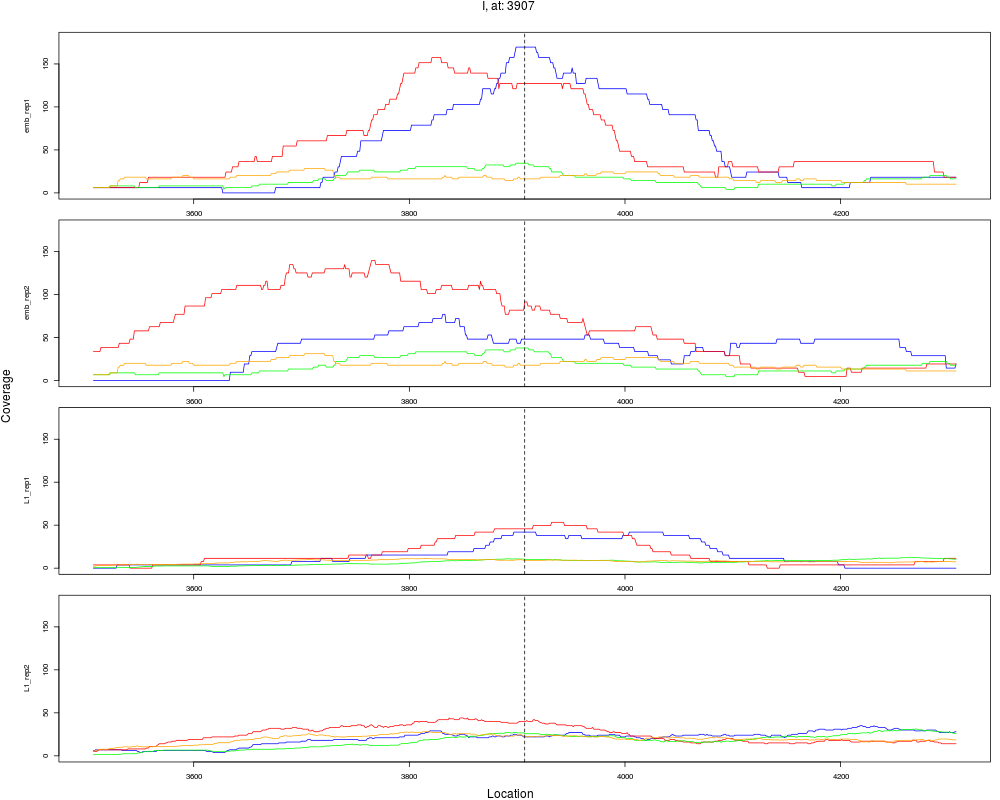
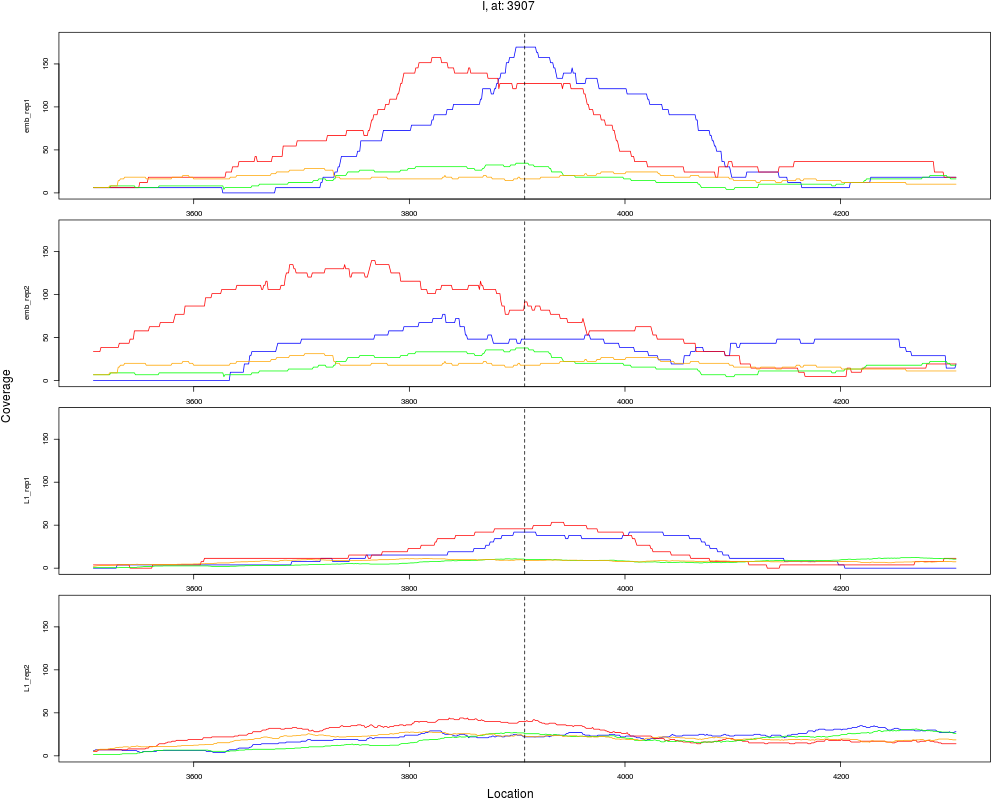
10 I 3907 1 emb 3907 0.3718658 7.035650e-04 3.377112e-03
> #pdf("Diff.Binding.pdf")
> plotPeak(rept, dat)
Warning message:
In rep(caption, length.out = nrow(rept)) :
'x' is NULL so the result will be NULL
> #dev.off()
>
> ## experienced users can proceed in a step by step fashion such that if program
> ## needs to be run for a different setting, intermediate results can be saved and reused.
> data("PHA4")
> conds <- factor(c("emb","emb","L1", "L1"), levels=c("emb", "L1"))
>
> bs.list <- read.binding.site.list(binding.site.list)
>
> ## compute consensus site
> consensus.site <- site.merge(bs.list, in.distance=100, out.distance=250)
merging sites from different conditions to consensus sites.done
>
> dat <- load.data(chip.data.list=chip.data.list, conds=conds, consensus.site=consensus.site, input.data.list=input.data.list, data.type="MCS")
reading data...done
computing normalization factor between ChIP and control samples.done
>
> ## count ChIP reads around each binding site
> dat <- get.site.count(dat, window.size=250)
count ChIP reads around each binding site.done
>
> ## test for differential binding
> dat <- test.diff.binding(dat)
Common dispersion: 0.05915362
>
> # report test result and plot the coverage profiles
> rept <- report.peak(dat)
> rept
chr pos nsig origin ori.pos FC.L1 pval FDR
1 I 260346 1 emb 260346 0.1129558 1.057645e-11 5.076698e-10
2 I 673122 1 emb 673122 0.1168590 1.798443e-10 4.316264e-09
3 I 757094 1 emb 757094 0.1717831 4.012809e-09 6.420495e-08
4 I 454904 1 emb 454904 0.1632294 1.349896e-08 1.619875e-07
5 I 547611 1 L1 547611 5.6827777 2.104486e-08 2.020307e-07
6 I 41410 1 L1 41410 4.2984821 4.548449e-07 3.638759e-06
7 I 546710 1 L1 546710 4.3025538 7.621600e-06 5.226240e-05
8 I 43116 1 emb 43116 0.2581921 6.466525e-05 3.879915e-04
9 I 159188 1 L1 159188 2.9888809 3.449015e-04 1.839475e-03
10 I 3907 1 emb 3907 0.3718658 7.035650e-04 3.377112e-03
> plotPeak(rept, dat)
Warning message:
In rep(caption, length.out = nrow(rept)) :
'x' is NULL so the result will be NULL
>
>
>
>
>
> dev.off()
null device
1
>
|