Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
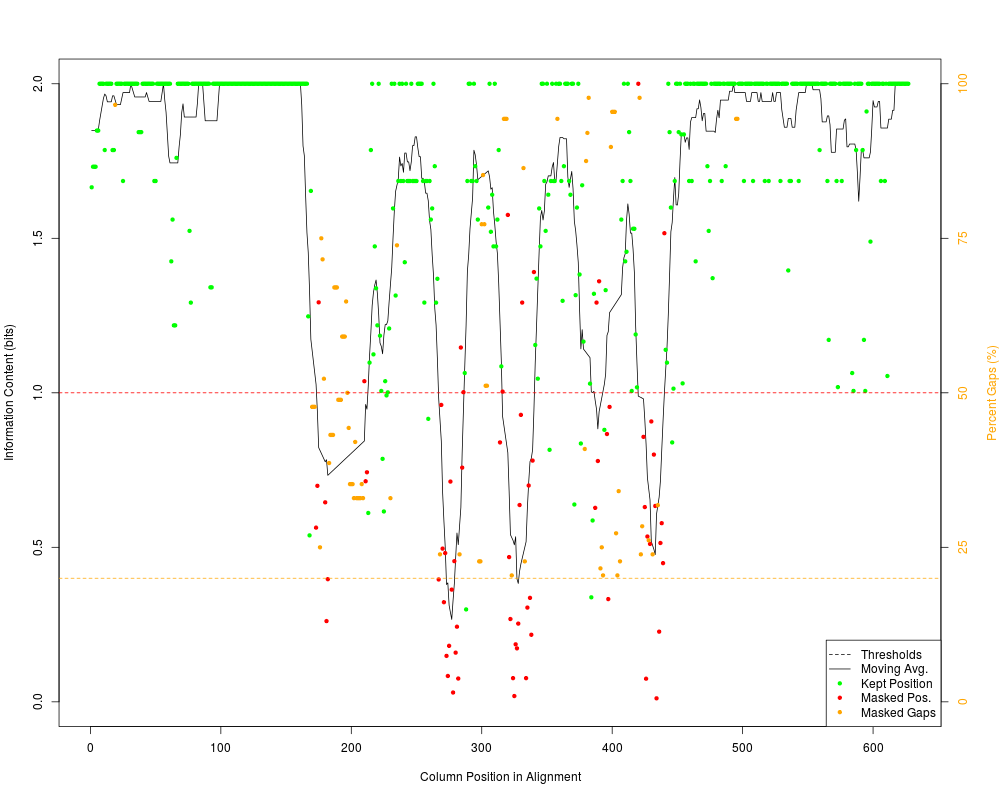
Mask Highly Variable Regions of An AlignmentDescriptionAutomatically masks poorly aligned regions of an alignment based on sequence conservation and gap frequency. Usage
MaskAlignment(myXStringSet,
windowSize = 5,
threshold = 1,
maxFractionGaps = 0.2,
showPlot = FALSE)
Arguments
DetailsPoorly aligned regions of a multiple sequence alignment may lead to incorrect results in downstream analyses, and require extra processing time. One method to mitigate their effects is to mask columns of the alignment that may be poorly aligned, such as highly-variable regions or regions with many insertions and deletions (gaps). Highly variable regions are detected by their signature of having low information content. A moving average of ValueA Author(s)Erik Wright DECIPHER@cae.wisc.edu See Also
Examples
fas <- system.file("extdata", "Streptomyces_ITS_aligned.fas", package="DECIPHER")
dna <- readDNAStringSet(fas)
masked_dna <- MaskAlignment(dna, showPlot=TRUE)
# display only unmasked nucleotides for use in downstream analyses
not_masked <- as(masked_dna, "DNAStringSet")
BrowseSeqs(not_masked)
# display only masked nucleotides that are covered by the mask
masked <- masked_dna
colmask(masked, append="replace", invert=TRUE) <- colmask(masked)
masked <- as(masked, "DNAStringSet")
BrowseSeqs(masked)
# display the complete DNA sequence set including the mask
masks <- lapply(width(colmask(masked_dna)), rep, x="+")
masks <- unlist(lapply(masks, paste, collapse=""))
masked_dna <- replaceAt(dna, at=IRanges(colmask(masked_dna)), value=masks)
BrowseSeqs(masked_dna)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(DECIPHER)
Loading required package: Biostrings
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Loading required package: RSQLite
Loading required package: DBI
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/DECIPHER/MaskAlignment.Rd_%03d_medium.png", width=480, height=480)
> ### Name: MaskAlignment
> ### Title: Mask Highly Variable Regions of An Alignment
> ### Aliases: MaskAlignment
>
> ### ** Examples
>
> fas <- system.file("extdata", "Streptomyces_ITS_aligned.fas", package="DECIPHER")
> dna <- readDNAStringSet(fas)
> masked_dna <- MaskAlignment(dna, showPlot=TRUE)
>
> # display only unmasked nucleotides for use in downstream analyses
> not_masked <- as(masked_dna, "DNAStringSet")
> BrowseSeqs(not_masked)
>
> # display only masked nucleotides that are covered by the mask
> masked <- masked_dna
> colmask(masked, append="replace", invert=TRUE) <- colmask(masked)
> masked <- as(masked, "DNAStringSet")
> BrowseSeqs(masked)
>
> # display the complete DNA sequence set including the mask
> masks <- lapply(width(colmask(masked_dna)), rep, x="+")
> masks <- unlist(lapply(masks, paste, collapse=""))
> masked_dna <- replaceAt(dna, at=IRanges(colmask(masked_dna)), value=masks)
> BrowseSeqs(masked_dna)
>
>
>
>
>
> dev.off()
null device
1
>
|