Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
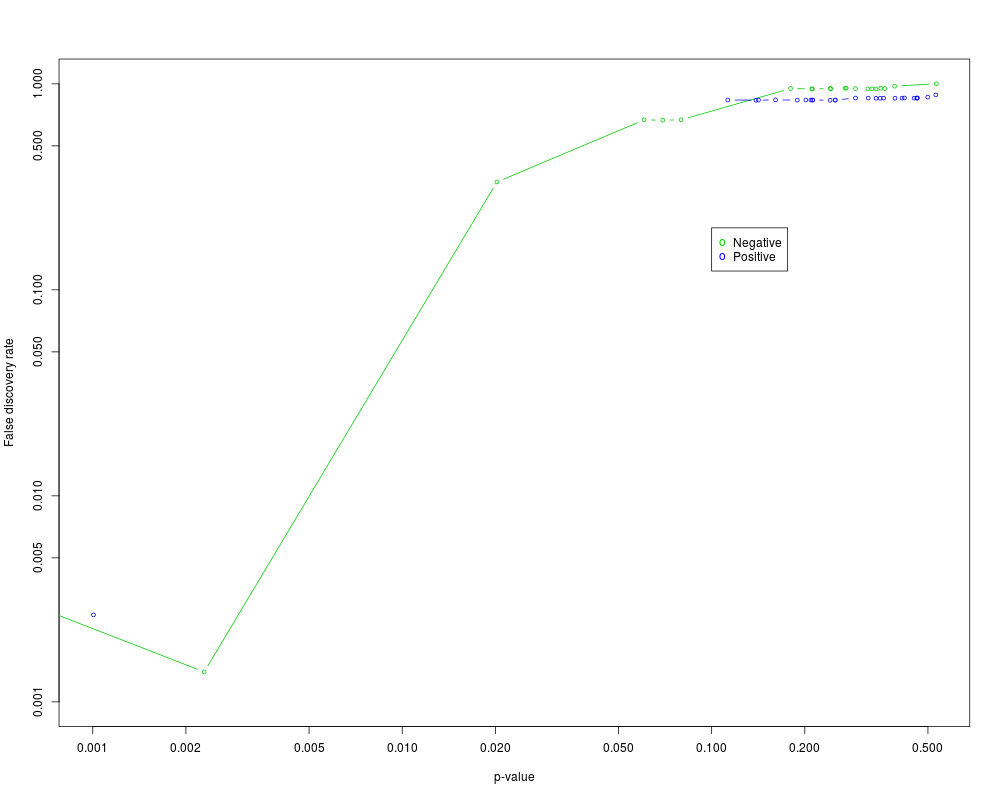
Plot the results from a Gene set analysisDescriptionPlots the results from a call to GSA (Gene set analysis) UsageGSA.plot(GSA.obj, fac=1, FDRcut = 1) Arguments
.
. DetailsThis function makes a plot of the significant gene sets, based on a call to the GSA (Gene set analysis) function. Author(s)Robert Tibshirani ReferencesEfron, B. and Tibshirani, R. On testing the significance of sets of genes. Stanford tech report rep 2006. http://www-stat.stanford.edu/~tibs/ftp/GSA.pdf Examples
######### two class unpaired comparison
# y must take values 1,2
set.seed(100)
x<-matrix(rnorm(1000*20),ncol=20)
dd<-sample(1:1000,size=100)
u<-matrix(2*rnorm(100),ncol=10,nrow=100)
x[dd,11:20]<-x[dd,11:20]+u
y<-c(rep(1,10),rep(2,10))
genenames=paste("g",1:1000,sep="")
#create some radnom gene sets
genesets=vector("list",50)
for(i in 1:50){
genesets[[i]]=paste("g",sample(1:1000,size=30),sep="")
}
geneset.names=paste("set",as.character(1:50),sep="")
GSA.obj<-GSA(x,y, genenames=genenames, genesets=genesets, resp.type="Two class unpaired", nperms=100)
GSA.listsets(GSA.obj, geneset.names=geneset.names,FDRcut=.5)
GSA.plot(GSA.obj)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(GSA)
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/GSA/GSA.plot.Rd_%03d_medium.png", width=480, height=480)
> ### Name: GSA.plot
> ### Title: Plot the results from a Gene set analysis
> ### Aliases: GSA.plot
> ### Keywords: univar survival ts nonparametric
>
> ### ** Examples
>
>
> ######### two class unpaired comparison
> # y must take values 1,2
>
> set.seed(100)
> x<-matrix(rnorm(1000*20),ncol=20)
> dd<-sample(1:1000,size=100)
>
> u<-matrix(2*rnorm(100),ncol=10,nrow=100)
> x[dd,11:20]<-x[dd,11:20]+u
> y<-c(rep(1,10),rep(2,10))
>
>
> genenames=paste("g",1:1000,sep="")
>
> #create some radnom gene sets
> genesets=vector("list",50)
> for(i in 1:50){
+ genesets[[i]]=paste("g",sample(1:1000,size=30),sep="")
+ }
> geneset.names=paste("set",as.character(1:50),sep="")
>
> GSA.obj<-GSA(x,y, genenames=genenames, genesets=genesets, resp.type="Two class unpaired", nperms=100)
perm= 10 / 100
perm= 20 / 100
perm= 30 / 100
perm= 40 / 100
perm= 50 / 100
perm= 60 / 100
perm= 70 / 100
perm= 80 / 100
perm= 90 / 100
perm= 100 / 100
>
>
> GSA.listsets(GSA.obj, geneset.names=geneset.names,FDRcut=.5)
$FDRcut
[1] 0.5
$negative
Gene_set Gene_set_name Score p-value FDR
[1,] "11" "set11" "-0.3151" "0" "0"
[2,] "15" "set15" "-0.6354" "0" "0"
[3,] "50" "set50" "-0.3587" "0.02" "0.3333"
$positive
Gene_set Gene_set_name Score p-value FDR
[1,] "6" "set6" "0.5037" "0" "0"
[2,] "17" "set17" "0.7162" "0" "0"
[3,] "31" "set31" "0.4192" "0" "0"
$nsets.neg
[1] 3
$nsets.pos
[1] 3
>
> GSA.plot(GSA.obj)
Warning messages:
1: In xy.coords(x, y, xlabel, ylabel, log) :
1 x value <= 0 omitted from logarithmic plot
2: In xy.coords(x, y, xlabel, ylabel, log) :
2 y values <= 0 omitted from logarithmic plot
>
>
>
>
>
>
> dev.off()
null device
1
>
|
Created & Maintained by Osamu Ogasawara (osamu.ogasawara@gmail.com) and 