Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
function to do genotypic quality controlDescriptionThis function helps selecting the marker which should enter into GWA analysis based on call rate, minor allele frequency, value of the chi-square test for Hardy-Weinberg equilibrium, and redudndance, defined as concordance between the distributions of the genotypes (including missing values). Usage
check.marker(data, snpsubset, idsubset, callrate = 0.95,
perid.call=0.95, extr.call = 0.1, extr.perid.call = 0.1, het.fdr = 0.01,
ibs.threshold = 0.95, ibs.mrk = 2000, ibs.exclude="both", maf, p.level = -1,
fdrate = 0.2, odds = 1000, hweidsubset, redundant = "no",
minconcordance = 2.0, qoption = "bh95", imphetasmissing = TRUE, XXY.call=0.8,
intermediateXF = c(0.5, 0.5))
Arguments
DetailsIn this procedure, sex errors are identified initally and then
possible residual errors are removed iteratively.
At the first step, of the iterative procedure,
per-marker (minor allele frequency, call rate,
exact P-value for Hardy-Weinberg equilibrium) and between-marker
statistics are generated and controlled for, mostly using the
internal call to the function At the second step of the iterative procedure,
per-person statistics, such call rate within
a person, heterozygosity and and between-person statistics
(identity by state across a random sample of markers) are generated,
using The procedure is applied recursively till no further markers and people are eliminated. ValueObject of class Author(s)Yurii Aulchenko See Also
Examples# usual way require(GenABEL.data) data(ge03d2c) # truncate the data to make the example faster ge03d2c <- ge03d2c[seq(from=1,to=nids(ge03d2c),by=2),seq(from=1,to=nsnps(ge03d2c),by=2)] # many errors mc0 <- check.marker(ge03d2c) # take only people and markers passing QC fixed0 <- ge03d2c[mc0$idok,mc0$snpok] # major errors fixed, still few males are heterozygous for X-chromsome markers mc1 <- check.marker(fixed0) # fix minor X-chromosome problems fixed1 <- Xfix(fixed0) # no errors mc2 <- check.marker(fixed1) summary(mc2) # ready to use fixed1 for analysis # let us look into redundancy require(GenABEL.data) data(srdta) mc <- check.marker(data=srdta,ids=c(1:300),call=.92,perid.call=.92) names(mc) mc$nohwe mc <- check.marker(data=srdta@gtdata[,1:100],call=0.95,perid.call=0.9, maf=0.02,minconcordance=0.9,fdr=0.1,redundant="all",ibs.mrk=0) summary(mc) HWE.show(data=srdta,snps=mc$nohwe) plot(mc) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(GenABEL)
Loading required package: MASS
Loading required package: GenABEL.data
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/GenABEL/check.marker.Rd_%03d_medium.png", width=480, height=480)
> ### Name: check.marker
> ### Title: function to do genotypic quality control
> ### Aliases: check.marker
> ### Keywords: misc
>
> ### ** Examples
>
> # usual way
> require(GenABEL.data)
> data(ge03d2c)
> # truncate the data to make the example faster
> ge03d2c <- ge03d2c[seq(from=1,to=nids(ge03d2c),by=2),seq(from=1,to=nsnps(ge03d2c),by=2)]
> # many errors
> mc0 <- check.marker(ge03d2c)
Excluding people/markers with extremely low call rate...
3795 markers and 98 people in total
0 people excluded because of call rate < 0.1
7 markers excluded because of call rate < 0.1
Passed: 3788 markers and 98 people
Running sex chromosome checks...
105 heterozygous X-linked male genotypes found
1 X-linked markers are likely to be autosomal (odds > 1000 )
1 male are likely to be female (odds > 1000 )
0 female are likely to be male (odds > 1000 )
0 people have intermediate X-chromosome inbreeding (0.5 > F > 0.5)
If these people/markers are removed, 1 heterozygous male genotypes are left
... these will be considered missing in analysis.
... Use Xfix() to fix these problems.
Passed: 3787 markers and 97 people
... 1 X/Y/mtDNA ( 1 0 0 ) impossible heterozygotes and female Ys set as missing
RUN 1
3787 markers and 97 people in total
424 (11.1962%) markers excluded as having low (<2.57732%) minor allele frequency
61 (1.610774%) markers excluded because of low (<95%) call rate
87 (2.297333%) markers excluded because they are out of HWE (FDR <0.2)
1 (1.030928%) people excluded because of low (<95%) call rate
Mean autosomal HET is 0.2862893 (s.e. 0.02788627)
1 (1.030928%) people excluded because too high autosomal heterozygosity (FDR <1%)
Excluded people had HET >= 0.4981387
Mean IBS is 0.760672 (s.e. 0.01904819), as based on 2000 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 3231 (85.31819%) markers passed all criteria
In total, 95 (97.93814%) people passed all criteria
RUN 2
3231 markers and 95 people in total
38 (1.176106%) markers excluded as having low (<2.631579%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<95%) call rate
Mean autosomal HET is 0.2872992 (s.e. 0.01758174)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7579581 (s.e. 0.0159936), as based on 2000 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 3193 (98.82389%) markers passed all criteria
In total, 95 (100%) people passed all criteria
RUN 3
3193 markers and 95 people in total
0 (0%) markers excluded as having low (<2.631579%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<95%) call rate
Mean autosomal HET is 0.2872992 (s.e. 0.01758174)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7577487 (s.e. 0.01657534), as based on 2000 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 3193 (100%) markers passed all criteria
In total, 95 (100%) people passed all criteria
> # take only people and markers passing QC
> fixed0 <- ge03d2c[mc0$idok,mc0$snpok]
> # major errors fixed, still few males are heterozygous for X-chromsome markers
> mc1 <- check.marker(fixed0)
Excluding people/markers with extremely low call rate...
3193 markers and 95 people in total
0 people excluded because of call rate < 0.1
0 markers excluded because of call rate < 0.1
Passed: 3193 markers and 95 people
Running sex chromosome checks...
1 heterozygous X-linked male genotypes found
0 X-linked markers are likely to be autosomal (odds > 1000 )
0 male are likely to be female (odds > 1000 )
0 female are likely to be male (odds > 1000 )
0 people have intermediate X-chromosome inbreeding (0.5 > F > 0.5)
If these people/markers are removed, 1 heterozygous male genotypes are left
... these will be considered missing in analysis.
... Use Xfix() to fix these problems.
Passed: 3193 markers and 95 people
... 1 X/Y/mtDNA ( 1 0 0 ) impossible heterozygotes and female Ys set as missing
RUN 1
3193 markers and 95 people in total
0 (0%) markers excluded as having low (<2.631579%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<95%) call rate
Mean autosomal HET is 0.2872992 (s.e. 0.01758174)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7602178 (s.e. 0.01609178), as based on 2000 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 3193 (100%) markers passed all criteria
In total, 95 (100%) people passed all criteria
> # fix minor X-chromosome problems
> fixed1 <- Xfix(fixed0)
... 1 X/Y/mtDNA ( 1 0 0 ) impossible heterozygotes and female Ys set as missing
> # no errors
> mc2 <- check.marker(fixed1)
Excluding people/markers with extremely low call rate...
3193 markers and 95 people in total
0 people excluded because of call rate < 0.1
0 markers excluded because of call rate < 0.1
Passed: 3193 markers and 95 people
Running sex chromosome checks...
0 heterozygous X-linked male genotypes found
0 X-linked markers are likely to be autosomal (odds > 1000 )
0 male are likely to be female (odds > 1000 )
0 female are likely to be male (odds > 1000 )
0 people have intermediate X-chromosome inbreeding (0.5 > F > 0.5)
If these people/markers are removed, 0 heterozygous male genotypes are left
Passed: 3193 markers and 95 people
no X/Y/mtDNA-errors to fix
RUN 1
3193 markers and 95 people in total
0 (0%) markers excluded as having low (<2.631579%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<95%) call rate
Mean autosomal HET is 0.2872992 (s.e. 0.01758174)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7601462 (s.e. 0.01642393), as based on 2000 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 3193 (100%) markers passed all criteria
In total, 95 (100%) people passed all criteria
> summary(mc2)
$`Per-SNP fails statistics`
NoCall NoMAF NoHWE Redundant Xsnpfail
NoCall 0 0 0 0 0
NoMAF NA 0 0 0 0
NoHWE NA NA 0 0 0
Redundant NA NA NA 0 0
Xsnpfail NA NA NA NA 0
$`Per-person fails statistics`
IDnoCall HetFail IBSFail isfemale ismale isXXY otherSexErr
IDnoCall 0 0 0 0 0 0 0
HetFail NA 0 0 0 0 0 0
IBSFail NA NA 0 0 0 0 0
isfemale NA NA NA 0 0 0 0
ismale NA NA NA NA 0 0 0
isXXY NA NA NA NA NA 0 0
otherSexErr NA NA NA NA NA NA 0
> # ready to use fixed1 for analysis
>
> # let us look into redundancy
> require(GenABEL.data)
> data(srdta)
> mc <- check.marker(data=srdta,ids=c(1:300),call=.92,perid.call=.92)
Excluding people/markers with extremely low call rate...
833 markers and 300 people in total
0 people excluded because of call rate < 0.1
0 markers excluded because of call rate < 0.1
Passed: 833 markers and 300 people
RUN 1
833 markers and 300 people in total
19 (2.280912%) markers excluded as having low (<0.8333333%) minor allele frequency
5 (0.6002401%) markers excluded because of low (<92%) call rate
2 (0.240096%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<92%) call rate
Mean autosomal HET is 0.3407554 (s.e. 0.04257531)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.729763 (s.e. 0.02863072), as based on 807 autosomal markers
2 (0.6666667%) people excluded because of too high IBS (>=0.95)
In total, 807 (96.87875%) markers passed all criteria
In total, 298 (99.33333%) people passed all criteria
RUN 2
807 markers and 298 people in total
0 (0%) markers excluded as having low (<0.8389262%) minor allele frequency
5 (0.6195787%) markers excluded because of low (<92%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<92%) call rate
Mean autosomal HET is 0.3406528 (s.e. 0.04267354)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7300679 (s.e. 0.02870589), as based on 802 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 802 (99.38042%) markers passed all criteria
In total, 298 (100%) people passed all criteria
RUN 3
802 markers and 298 people in total
0 (0%) markers excluded as having low (<0.8389262%) minor allele frequency
0 (0%) markers excluded because of low (<92%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.2)
0 (0%) people excluded because of low (<92%) call rate
Mean autosomal HET is 0.3406528 (s.e. 0.04267354)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
Mean IBS is 0.7300679 (s.e. 0.02870589), as based on 802 autosomal markers
0 (0%) people excluded because of too high IBS (>=0.95)
In total, 802 (100%) markers passed all criteria
In total, 298 (100%) people passed all criteria
> names(mc)
[1] "nofreq" "nocall" "nohwe" "idnocall" "ibsfail" "snpok" "idok"
[8] "call"
> mc$nohwe
[1] "rs73" "rs128"
> mc <- check.marker(data=srdta@gtdata[,1:100],call=0.95,perid.call=0.9,
+ maf=0.02,minconcordance=0.9,fdr=0.1,redundant="all",ibs.mrk=0)
Excluding people/markers with extremely low call rate...
100 markers and 2500 people in total
0 people excluded because of call rate < 0.1
0 markers excluded because of call rate < 0.1
Passed: 100 markers and 2500 people
RUN 1
100 markers and 2500 people in total
5 (5%) markers excluded as having low (<2%) minor allele frequency
37 (37%) markers excluded because of low (<95%) call rate
2 (2%) markers excluded because they are out of HWE (FDR <0.1)
48 (1.92%) people excluded because of low (<90%) call rate
Mean autosomal HET is 0.3542481 (s.e. 0.1151198)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
In total, 60 (60%) markers passed all criteria
In total, 2452 (98.08%) people passed all criteria
RUN 2
60 markers and 2452 people in total
0 (0%) markers excluded as having low (<2%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.1)
0 (0%) people excluded because of low (<90%) call rate
Mean autosomal HET is 0.3542175 (s.e. 0.1151843)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
In total, 60 (100%) markers passed all criteria
In total, 2452 (100%) people passed all criteria
CHECK REDUNDANCY
60 markers and 2452 people in total
5 (8.333333%) markers excluded as redundant (option = "all")
0 (0%) markers excluded as having low (<2%) minor allele frequency
0 (0%) markers excluded because of low (<95%) call rate
0 (0%) markers excluded because they are out of HWE (FDR <0.1)
44 (1.794454%) people excluded because of low (<90%) call rate
Mean autosomal HET is 0.3529672 (s.e. 0.1107675)
0 people excluded because too high autosomal heterozygosity (FDR <1%)
In total, 55 (91.66667%) markers passed all criteria
In total, 2408 (98.20555%) people passed all criteria
> summary(mc)
$`Per-SNP fails statistics`
NoCall NoMAF NoHWE Redundant Xsnpfail
NoCall 34 3 0 0 0
NoMAF NA 1 1 0 0
NoHWE NA NA 1 0 0
Redundant NA NA NA 5 0
Xsnpfail NA NA NA NA 0
$`Per-person fails statistics`
IDnoCall HetFail IBSFail isfemale ismale isXXY otherSexErr
IDnoCall 92 0 0 0 0 0 0
HetFail NA 0 0 0 0 0 0
IBSFail NA NA 0 0 0 0 0
isfemale NA NA NA 0 0 0 0
ismale NA NA NA NA 0 0 0
isXXY NA NA NA NA NA 0 0
otherSexErr NA NA NA NA NA NA 0
> HWE.show(data=srdta,snps=mc$nohwe)
HWE summary for rs73 :
A/A A/B B/B
observed 2331.0000000 44.00000 10.0000000
expected 2321.4293501 63.14130 0.4293501
chi2+ 0.0394573 5.80269 213.3395064
Chi2 = 219.1817 ; P = 1.364229e-49; exact P = 1.79247e-12
HWE summary for rs128 :
A/A A/B B/B
observed 2.281000e+03 101.000000 9.000000
expected 2.273481e+03 116.038687 1.480657
chi2+ 2.486959e-02 1.949023 38.186115
Chi2 = 40.16001 ; P = 2.339897e-10; exact P = 9.408599e-06
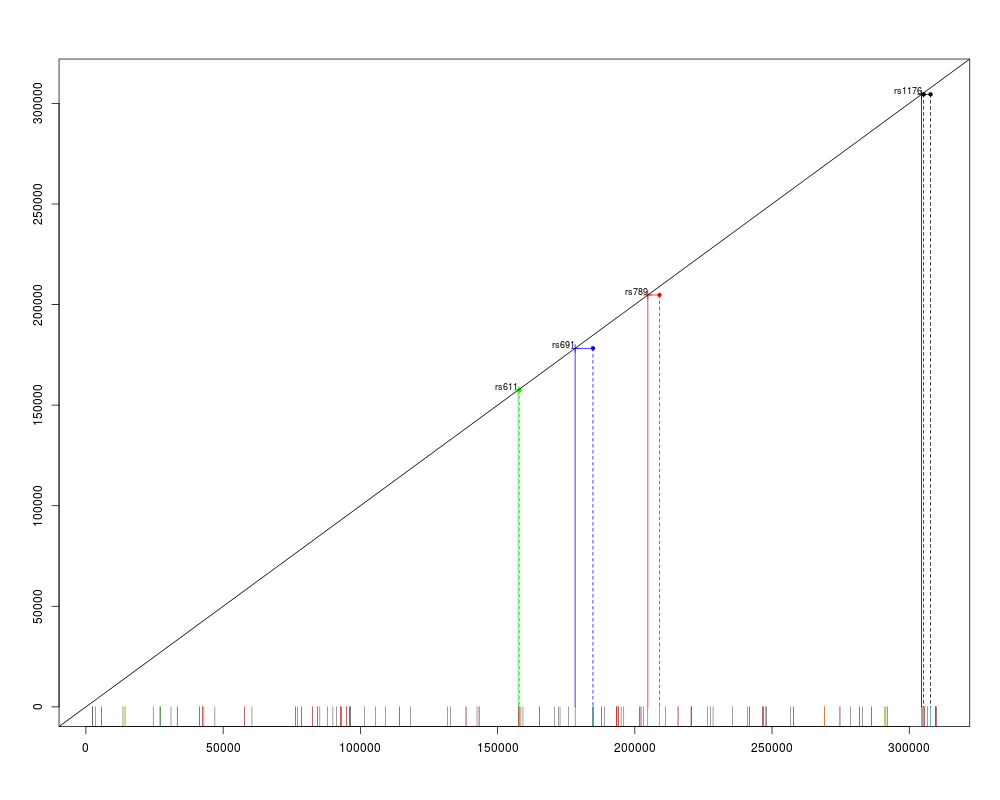
> plot(mc)
Red: no call
Yellow: low frequency
Green: out of HWE
Cyan: redundant
Diagonal: redundant markers (reference presented as "+")
>
>
>
>
>
>
> dev.off()
null device
1
>
|