Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Highest (Posterior) Density IntervalDescriptionCalculate the highest density interval (HDI) for a probability distribution for a given probability mass. This is often applied to a Bayesian posterior distribution and is then termed "highest posterior density interval", but can be applied to any distribution, including priors. The function is an S3 generic, with methods for a range of input objects. Usagehdi(object, credMass = 0.95, ...) ## Default S3 method: hdi(object, credMass = 0.95, ...) ## S3 method for class 'function' hdi(object, credMass = 0.95, tol, ...) ## S3 method for class 'matrix' hdi(object, credMass = 0.95, ...) ## S3 method for class 'data.frame' hdi(object, credMass = 0.95, ...) ## S3 method for class 'list' hdi(object, credMass = 0.95, ...) ## S3 method for class 'density' hdi(object, credMass = 0.95, allowSplit=FALSE, ...) ## S3 method for class 'mcmc' hdi(object, credMass = 0.95, ...) ## S3 method for class 'mcmc.list' hdi(object, credMass = 0.95, ...) ## S3 method for class 'mcarray' hdi(object, credMass = 0.95, ...) ## S3 method for class 'bugs' hdi(object, credMass = 0.95, ...) ## S3 method for class 'jagsUI' hdi(object, credMass = 0.95, ...) ## S3 method for class 'rjags' hdi(object, credMass = 0.95, ...) ## S3 method for class 'runjags' hdi(object, credMass = 0.95, ...) Arguments
DetailsThe HDI is the interval which contains the required mass such that all points within the interval have a higher probability density than points outside the interval.
In contrast, a symmetric density interval defined by (eg.) the 10% and 90% quantiles may include values with lower probability than those excluded. For a distribution that is not severely multimodal, the HDI is the narrowest interval containing the specified mass, and the
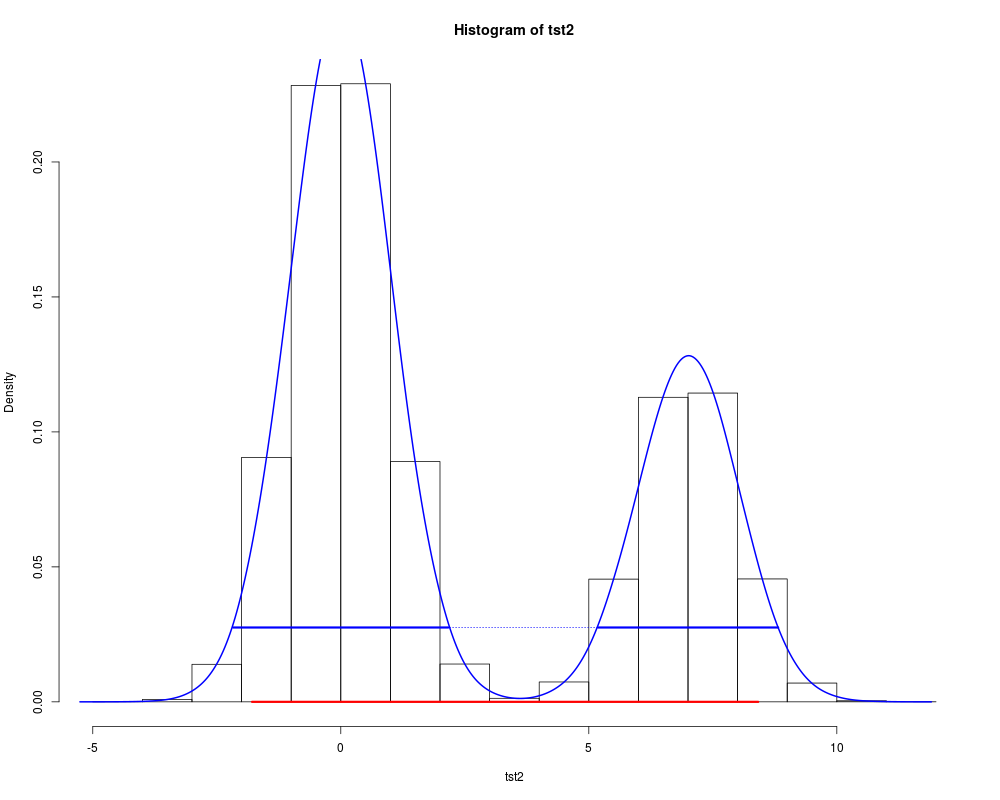
The default method expects a vector representing draws from the target distribution, such as is produced by an MCMC process. Missing values are silently ignored; if the vector has no non-missing values, NAs are returned. The The The The packages R2winbugs and R2openbugs produce None of the above use interpolation: the values returned correspond to specific values in the data object, and will be conservative (ie, too wide rather than too narrow). Results thus depend on the random draws, and will be unstable if few values are provided. For a 95% HDI, 10,000 independent draws are recommended; a smaller number will be adequate for a 80% HDI, many more for a 99% HDI. The function method requires the name for the inverse cumulative density function (ICDF) of the distribution; standard R functions for this have a Valuea vector of length 2 or a 2-row matrix with the lower and upper limits of the HDI, with an attribute "credMass". The Author(s)Mike Meredith and John Krushke. Code for ReferencesKruschke, J. K. 2011. Doing Bayesian data analysis: a tutorial with R and BUGS. Elsevier, Amsterdam, section 3.3.5. Examples# for a vector: tst <- rgamma(1e5, 2.5, 2) hdi(tst) hdi(tst, credMass=0.8) # For comparison, the symmetrical 80% CrI: quantile(tst, c(0.1,0.9)) # for a density: dens <- density(tst) hdi(dens, credMass=0.8) # Now a data frame: tst <- data.frame(mu = rnorm(1e4, 4, 1), sigma = rlnorm(1e4)) hdi(tst, 0.8) apply(tst, 2, quantile, c(0.1,0.9)) tst$txt <- LETTERS[1:25] hdi(tst, 0.8) # For a function: hdi(qgamma, 0.8, shape=2.5, rate=2) # and the symmetrical 80% CrI: qgamma(c(0.1, 0.9), 2.5, 2) # A severely bimodal distribution: tst2 <- c(rnorm(1e5), rnorm(5e4, 7)) hist(tst2, freq=FALSE) (hdiMC <- hdi(tst2)) segments(hdiMC[1], 0, hdiMC[2], 0, lwd=3, col='red') # This is a valid 95% CrI, but not a Highest Density Interval dens2 <- density(tst2) lines(dens2, lwd=2, col='blue') (hdiD1 <- hdi(dens2)) # default allowSplit = FALSE; note the warning (ht <- attr(hdiD1, "height")) segments(hdiD1[1], ht, hdiD1[2], ht, lty=3, col='blue') (hdiD2 <- hdi(dens2, allowSplit=TRUE)) segments(hdiD2[, 1], ht, hdiD2[, 2], ht, lwd=3, col='blue') # This is the correct 95% HDI. Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(HDInterval)
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/HDInterval/hdi.Rd_%03d_medium.png", width=480, height=480)
> ### Name: hdi
> ### Title: Highest (Posterior) Density Interval
> ### Aliases: HDInterval hdi hdi.default hdi.function hdi.matrix
> ### hdi.data.frame hdi.list hdi.density hdi.mcmc hdi.mcarray
> ### hdi.mcmc.list hdi.bugs hdi.jagsUI hdi.rjags hdi.runjags
> ### Keywords: methods htest
>
> ### ** Examples
>
> # for a vector:
> tst <- rgamma(1e5, 2.5, 2)
> hdi(tst)
lower upper
0.07421291 2.77720536
attr(,"credMass")
[1] 0.95
> hdi(tst, credMass=0.8)
lower upper
0.1870523 1.8901953
attr(,"credMass")
[1] 0.8
> # For comparison, the symmetrical 80% CrI:
> quantile(tst, c(0.1,0.9))
10% 90%
0.4056557 2.3024642
>
> # for a density:
> dens <- density(tst)
> hdi(dens, credMass=0.8)
lower upper
0.206056 1.913838
attr(,"credMass")
[1] 0.8
attr(,"height")
[1] 0.2393585
>
> # Now a data frame:
> tst <- data.frame(mu = rnorm(1e4, 4, 1), sigma = rlnorm(1e4))
> hdi(tst, 0.8)
mu sigma
lower 2.776759 0.06131656
upper 5.358374 2.30314266
attr(,"credMass")
[1] 0.8
> apply(tst, 2, quantile, c(0.1,0.9))
mu sigma
10% 2.726329 0.2721758
90% 5.314381 3.6120315
> tst$txt <- LETTERS[1:25]
> hdi(tst, 0.8)
mu sigma txt
lower 2.776759 0.06131656 NA
upper 5.358374 2.30314266 NA
attr(,"credMass")
[1] 0.8
>
> # For a function:
> hdi(qgamma, 0.8, shape=2.5, rate=2)
lower upper
0.1947158 1.9053925
attr(,"credMass")
[1] 0.8
> # and the symmetrical 80% CrI:
> qgamma(c(0.1, 0.9), 2.5, 2)
[1] 0.402577 2.309089
>
> # A severely bimodal distribution:
> tst2 <- c(rnorm(1e5), rnorm(5e4, 7))
> hist(tst2, freq=FALSE)
> (hdiMC <- hdi(tst2))
lower upper
-1.835687 8.378939
attr(,"credMass")
[1] 0.95
> segments(hdiMC[1], 0, hdiMC[2], 0, lwd=3, col='red')
> # This is a valid 95% CrI, but not a Highest Density Interval
>
> dens2 <- density(tst2)
> lines(dens2, lwd=2, col='blue')
> (hdiD1 <- hdi(dens2)) # default allowSplit = FALSE; note the warning
lower upper
-1.916874 8.462495
attr(,"credMass")
[1] 0.95
attr(,"height")
[1] 0.02838018
Warning message:
In hdi.density(dens2) : The HDI is discontinuous but allowSplit = FALSE;
the result is a valid CrI but not HDI.
> (ht <- attr(hdiD1, "height"))
[1] 0.02838018
> segments(hdiD1[1], ht, hdiD1[2], ht, lty=3, col='blue')
> (hdiD2 <- hdi(dens2, allowSplit=TRUE))
begin end
[1,] -2.172105 2.183828
[2,] 5.212562 8.819818
attr(,"credMass")
[1] 0.95
attr(,"height")
[1] 0.02838018
> segments(hdiD2[, 1], ht, hdiD2[, 2], ht, lwd=3, col='blue')
> # This is the correct 95% HDI.
>
>
>
>
>
> dev.off()
null device
1
>
|