Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Samples gene family trees that evolved on a species tree with various conditions.DescriptionSamples gene family trees that evolved on the branches of a species tree under the birth-death process. Trees may be conditioned on various priors for the number of reconstructed gene lineages at the root of the species tree, on the total number of genes in the gene tree, on the number of genes in each tip of the species tree, on at least one gene in each tip of the species tree or some combination of those conditions. Usagergenetree(n, spec.phy, lams, mus, root = NULL, genetips = NULL, alltips = FALSE) Arguments
. Details
ValueIf Author(s)Nathaniel Malachi Hallinan ReferencesN. Hallinan. Null models for gene family trees, Math. Biosci. (In review). See Also
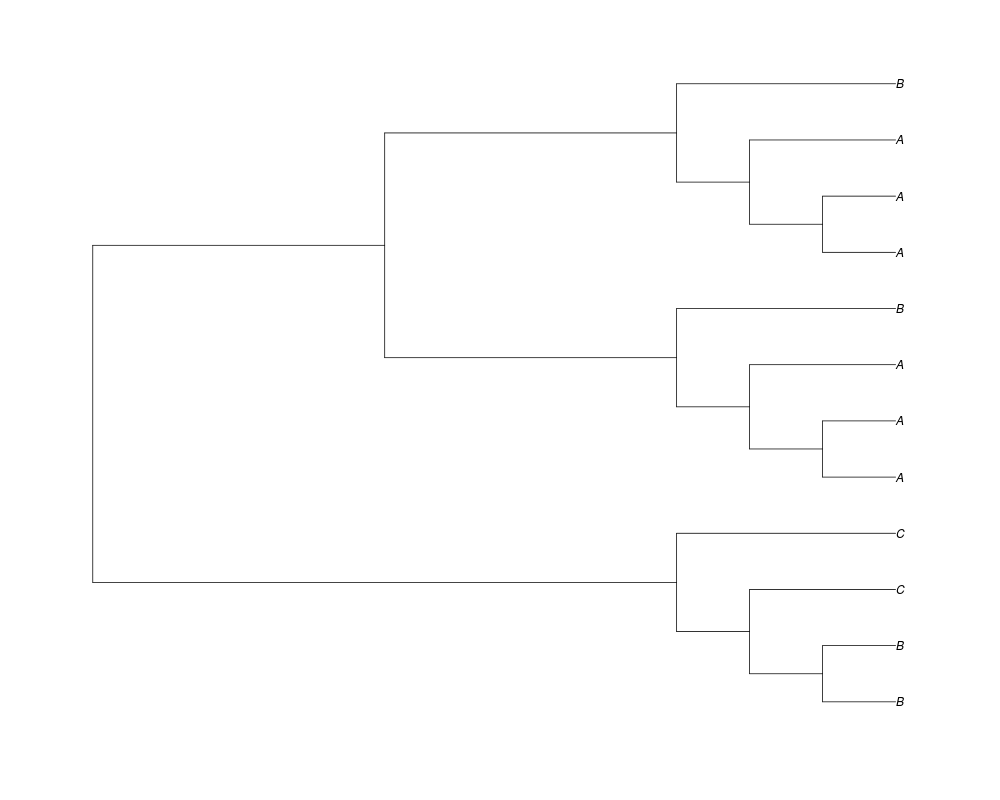
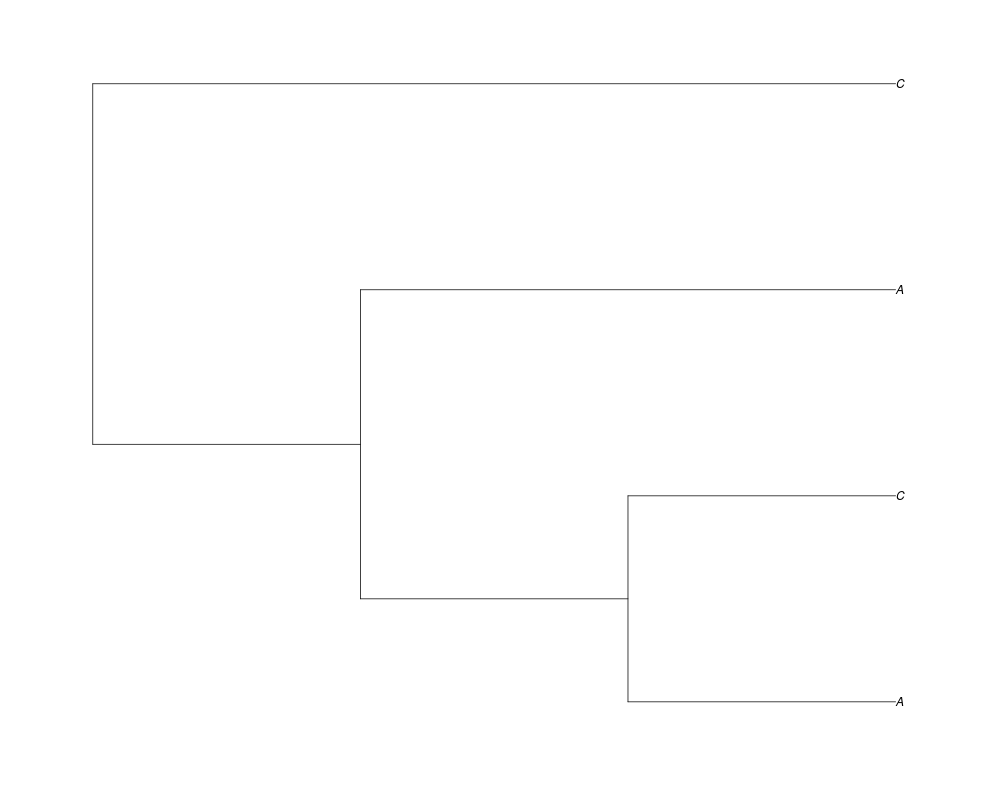
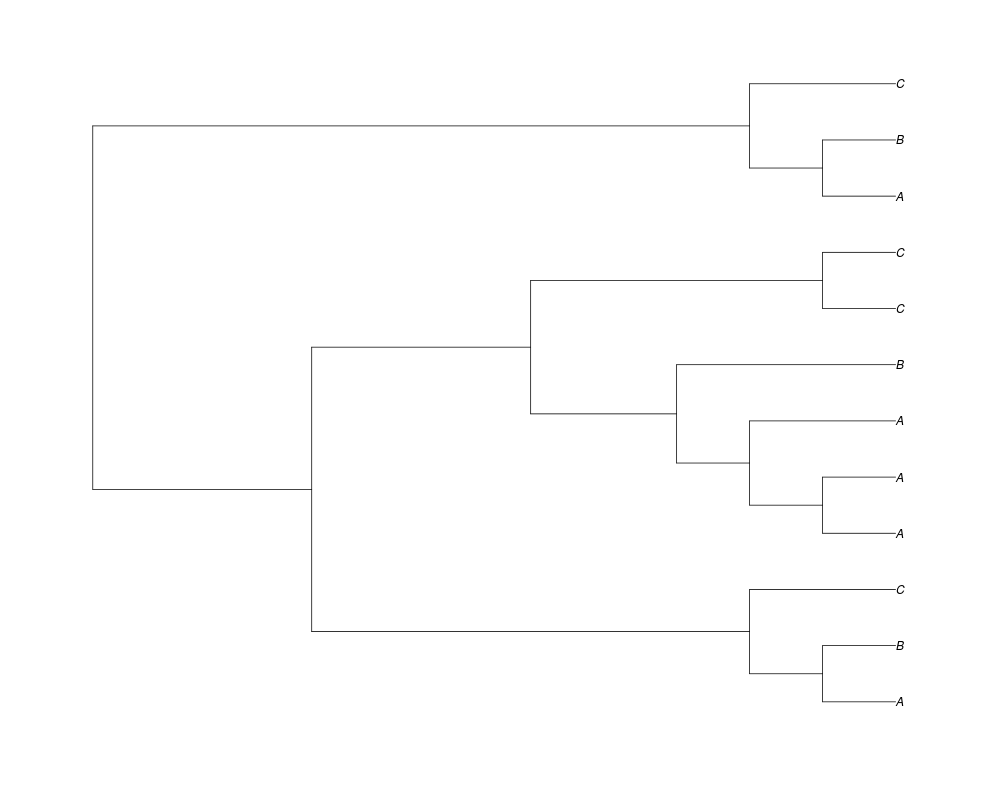
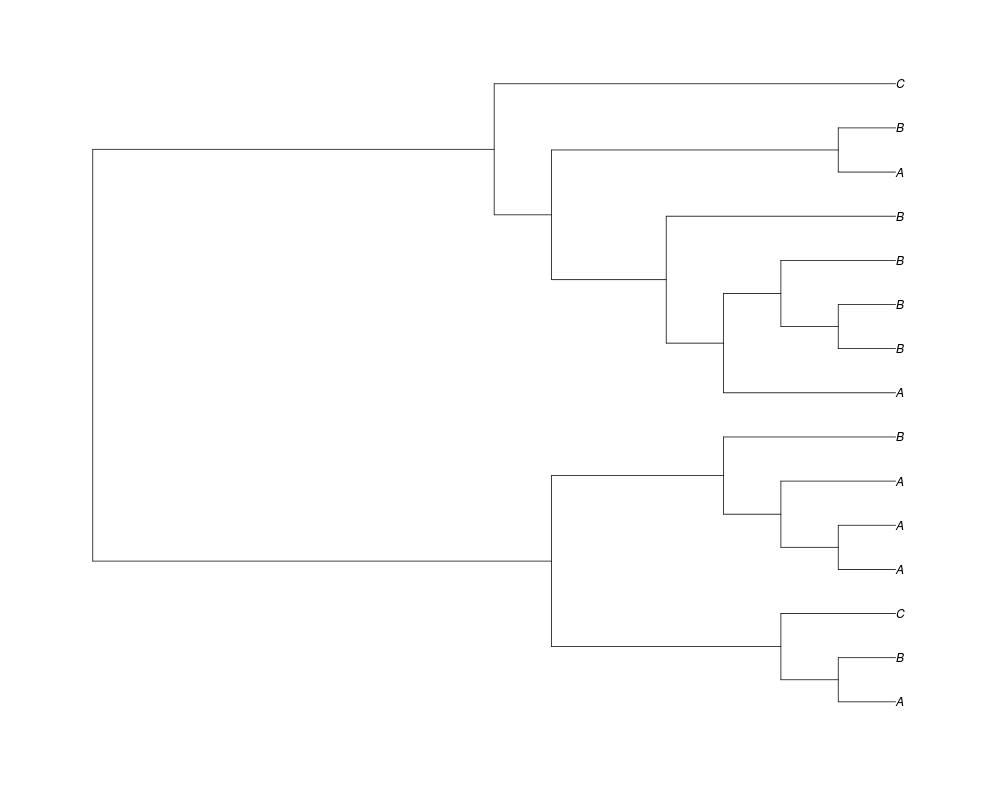
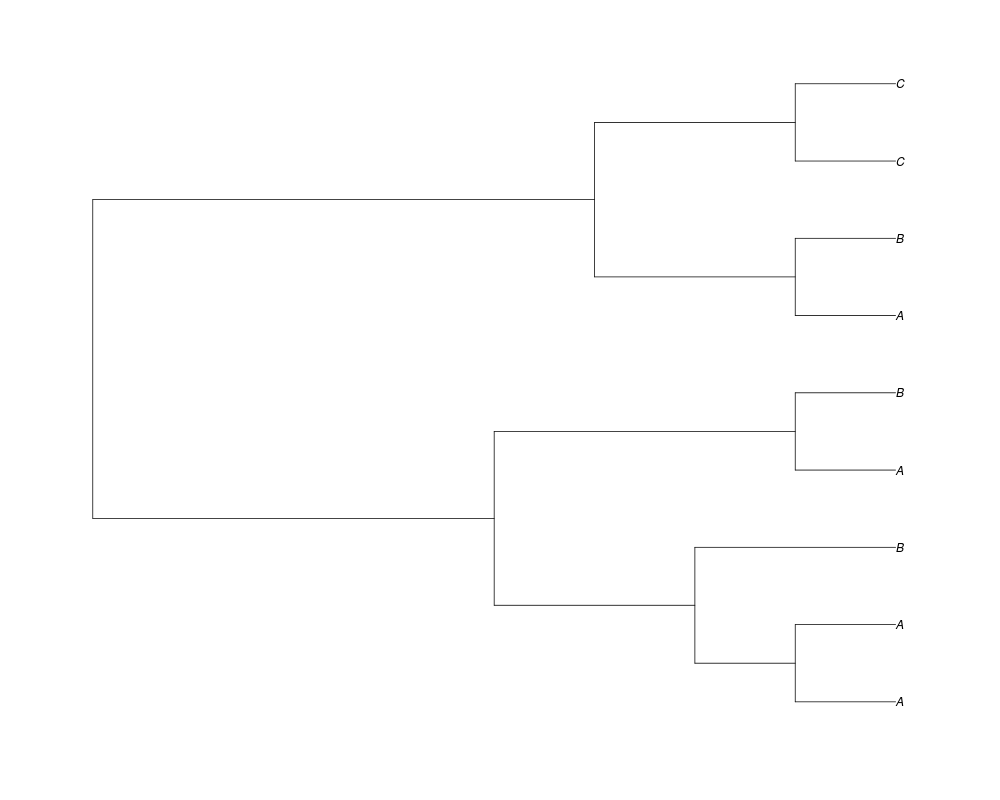
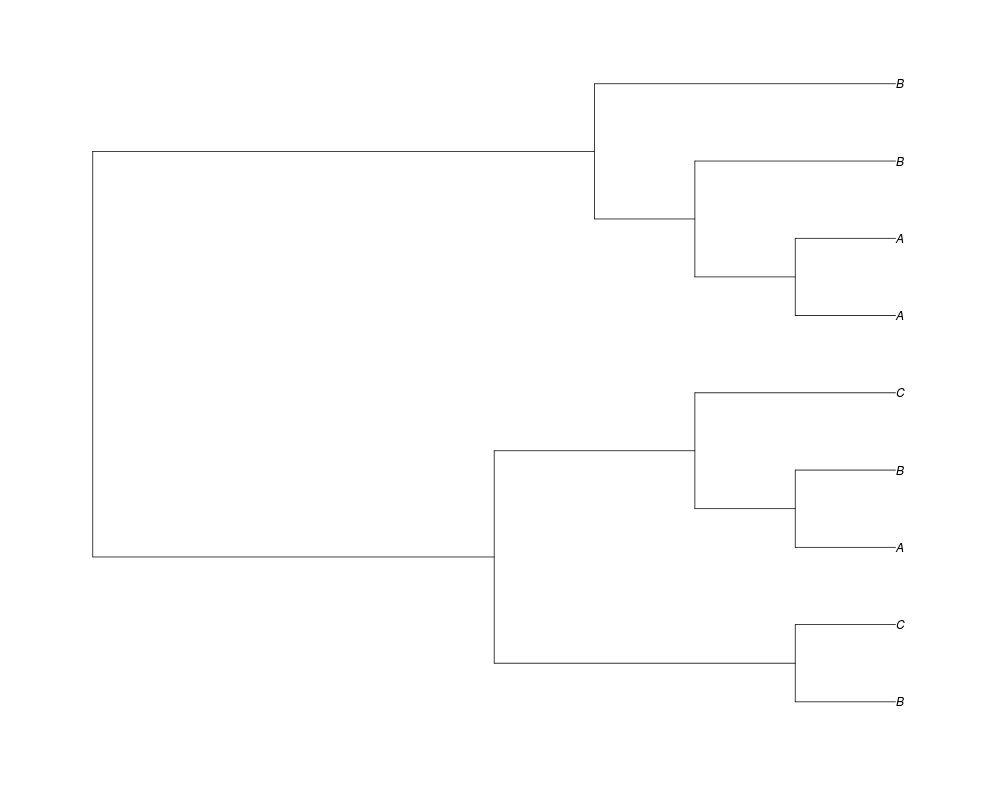
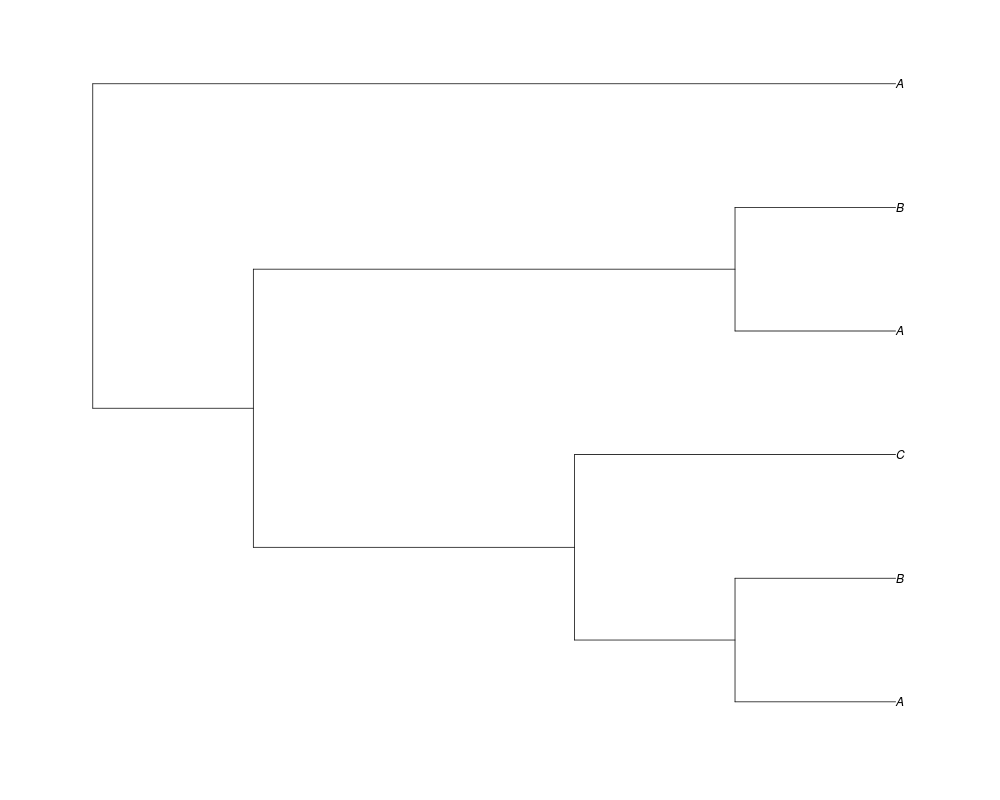
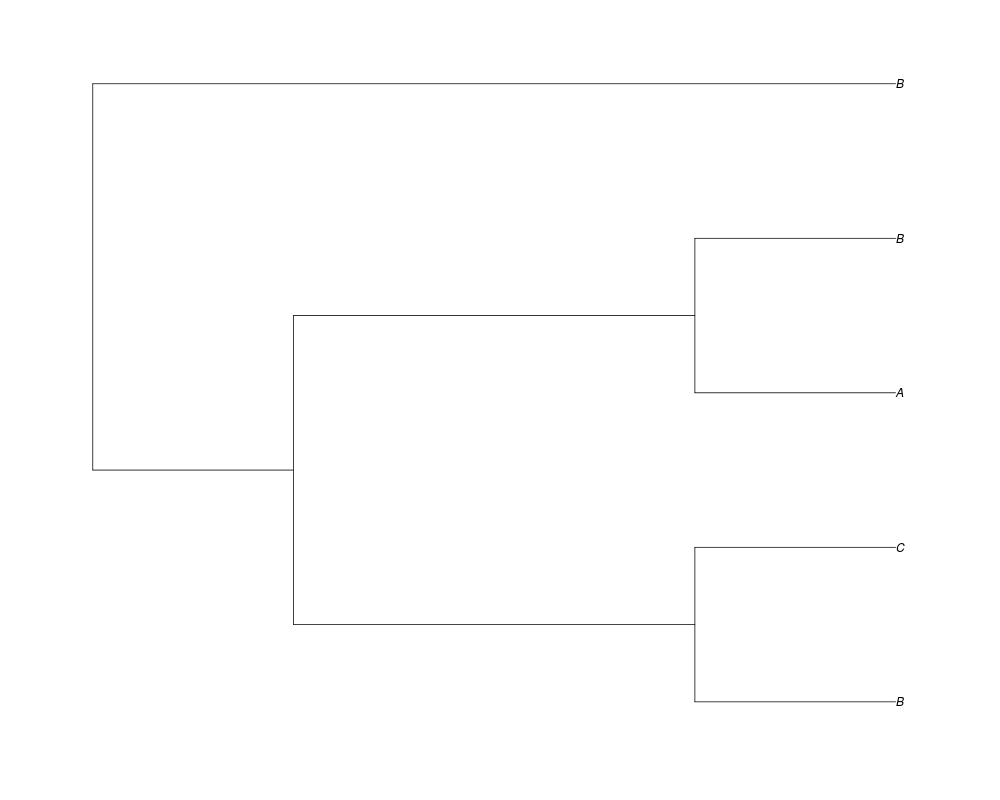
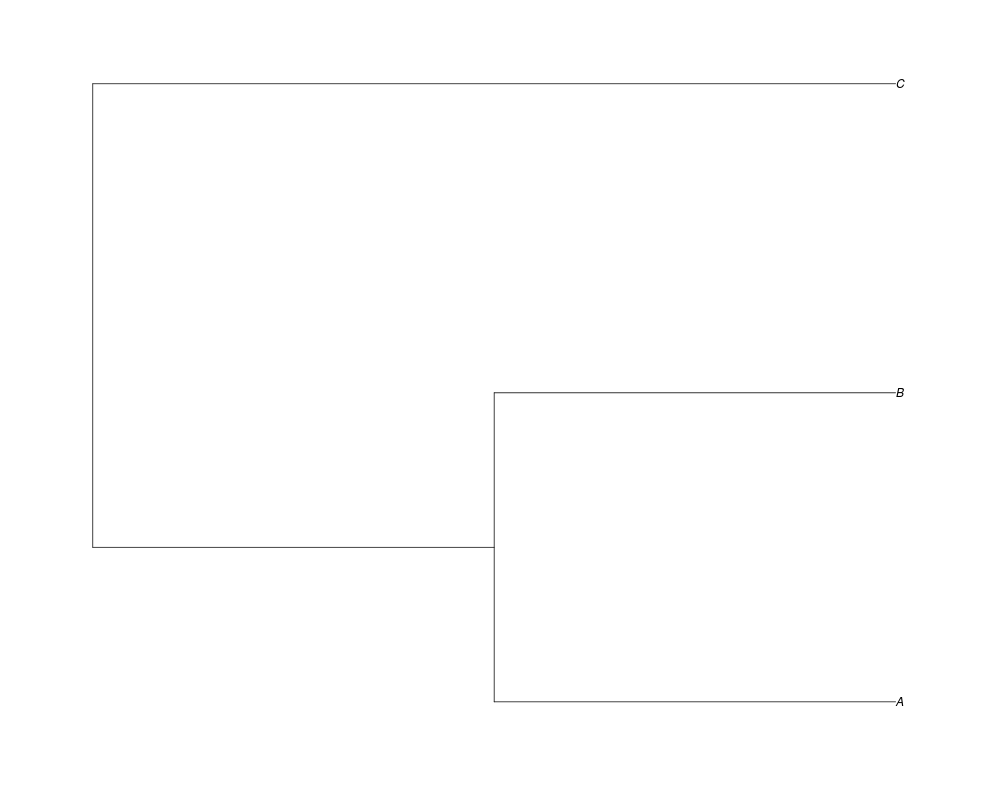
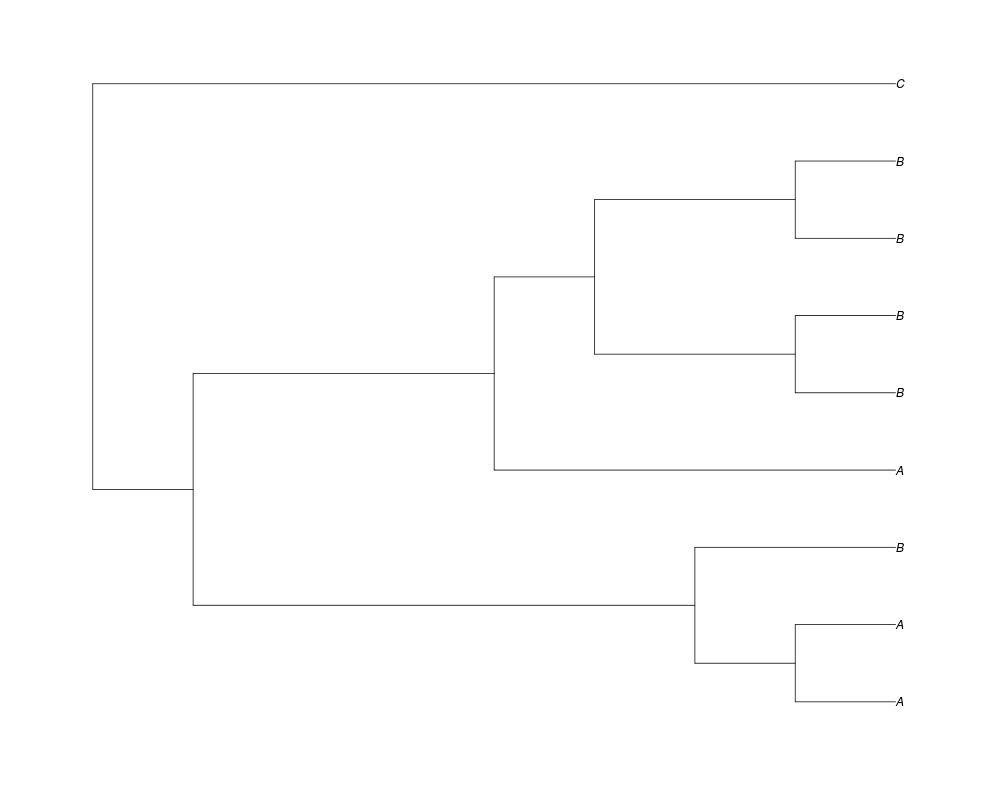
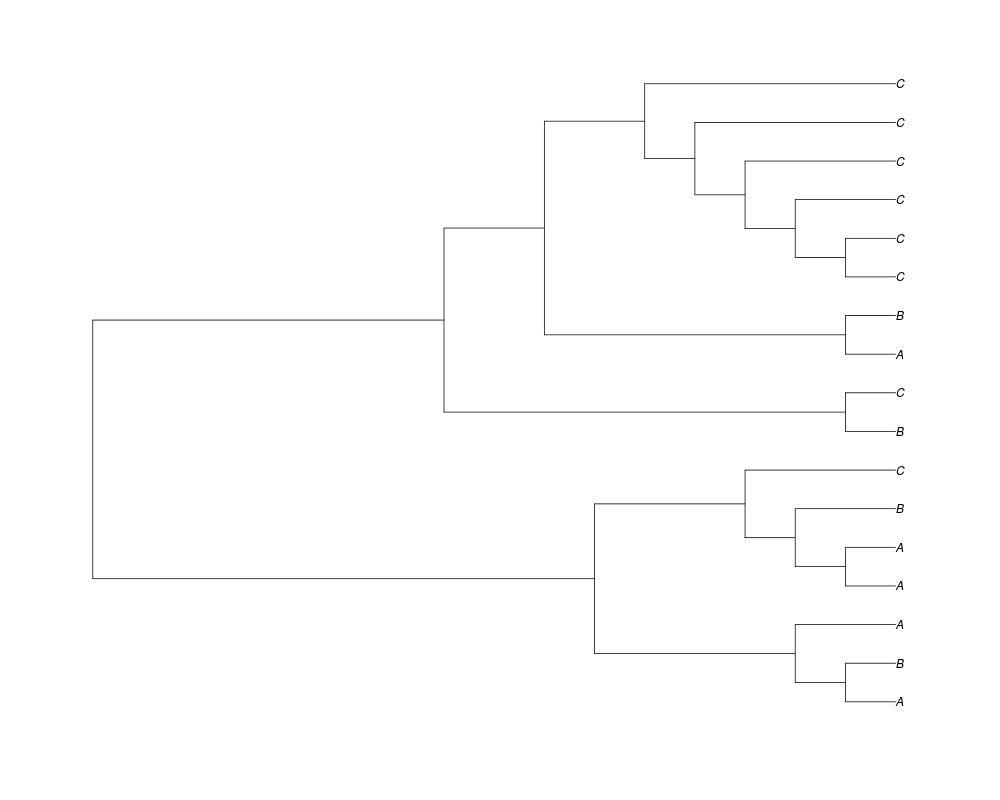
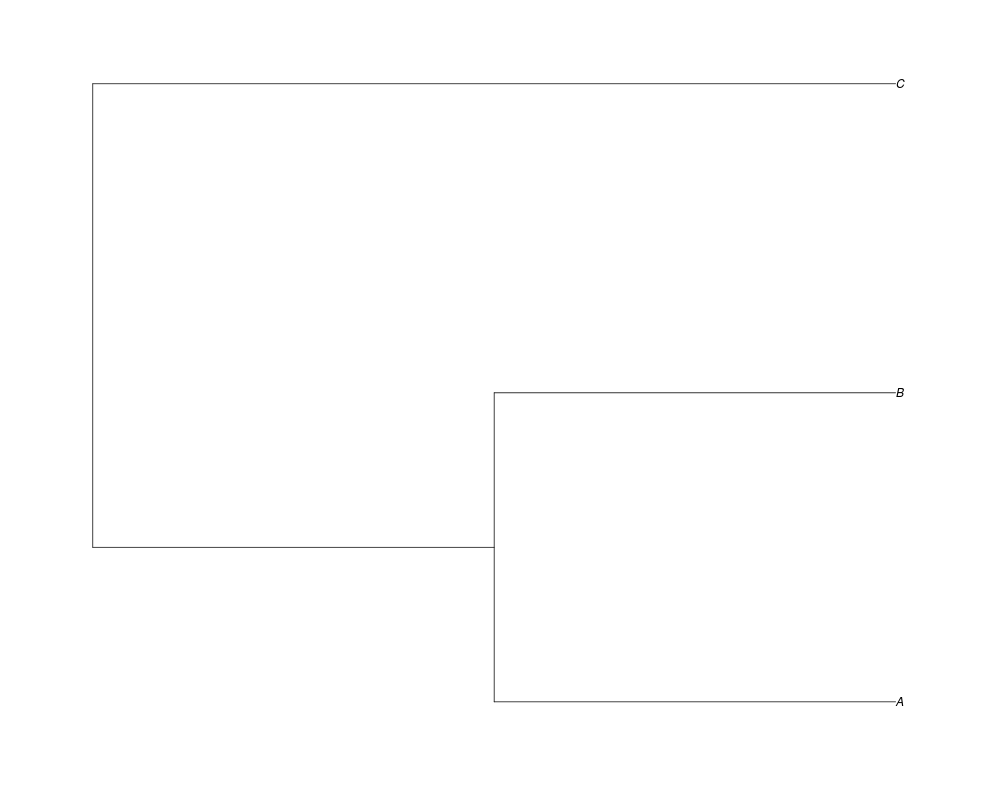
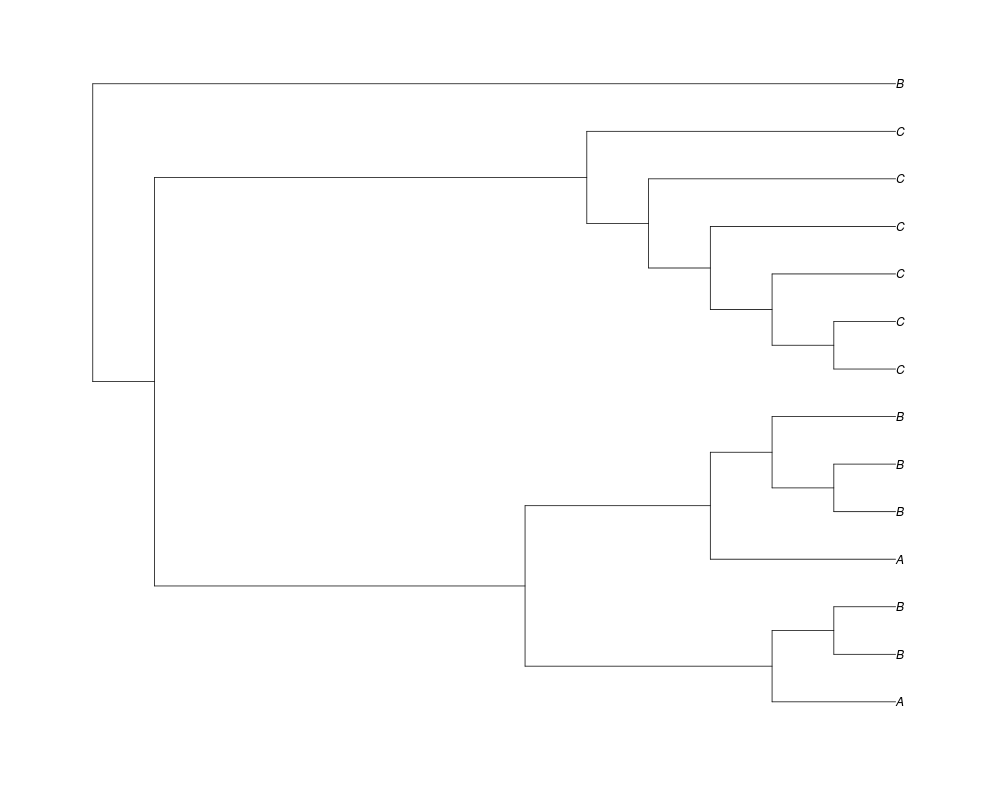
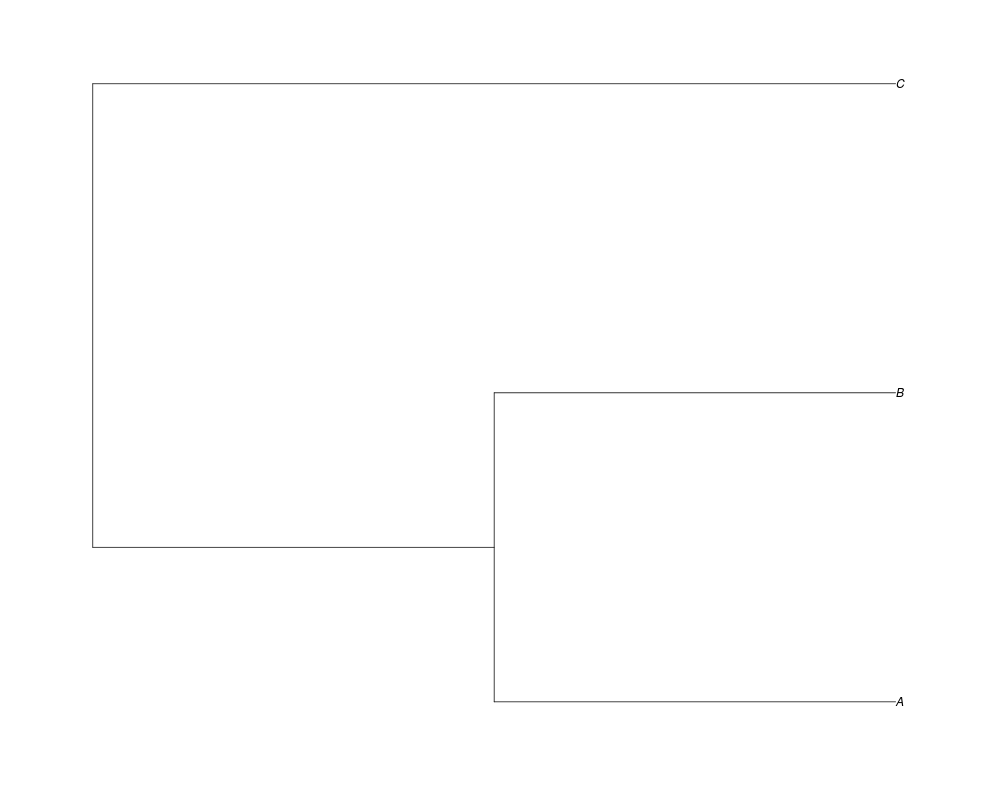
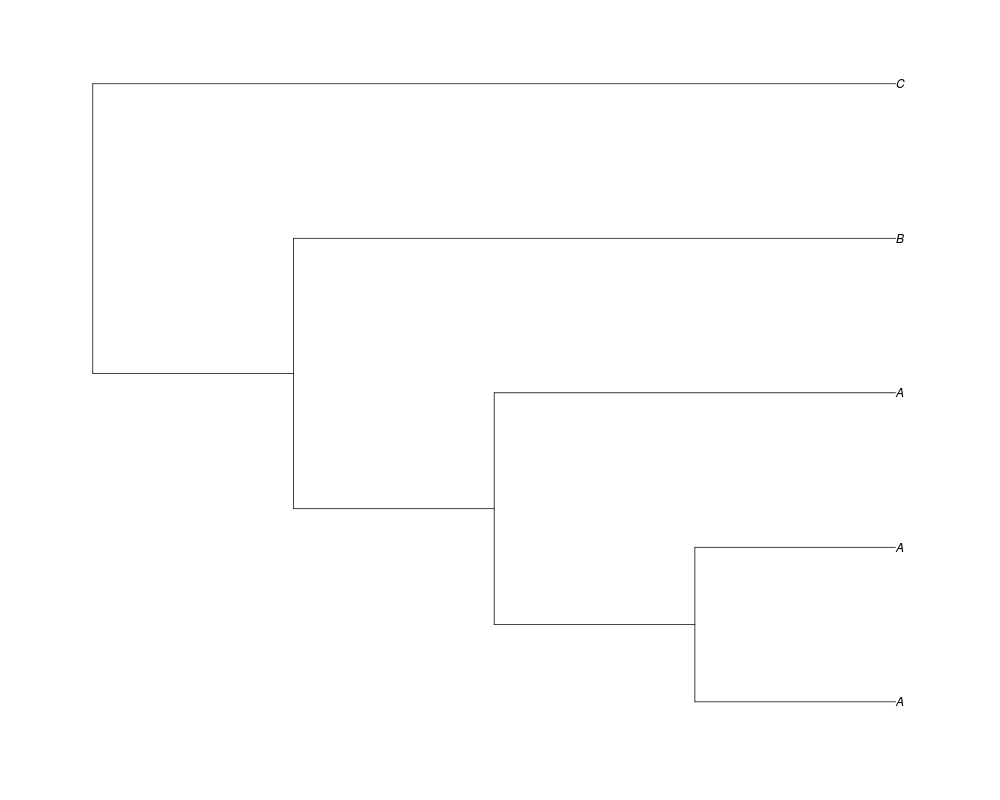
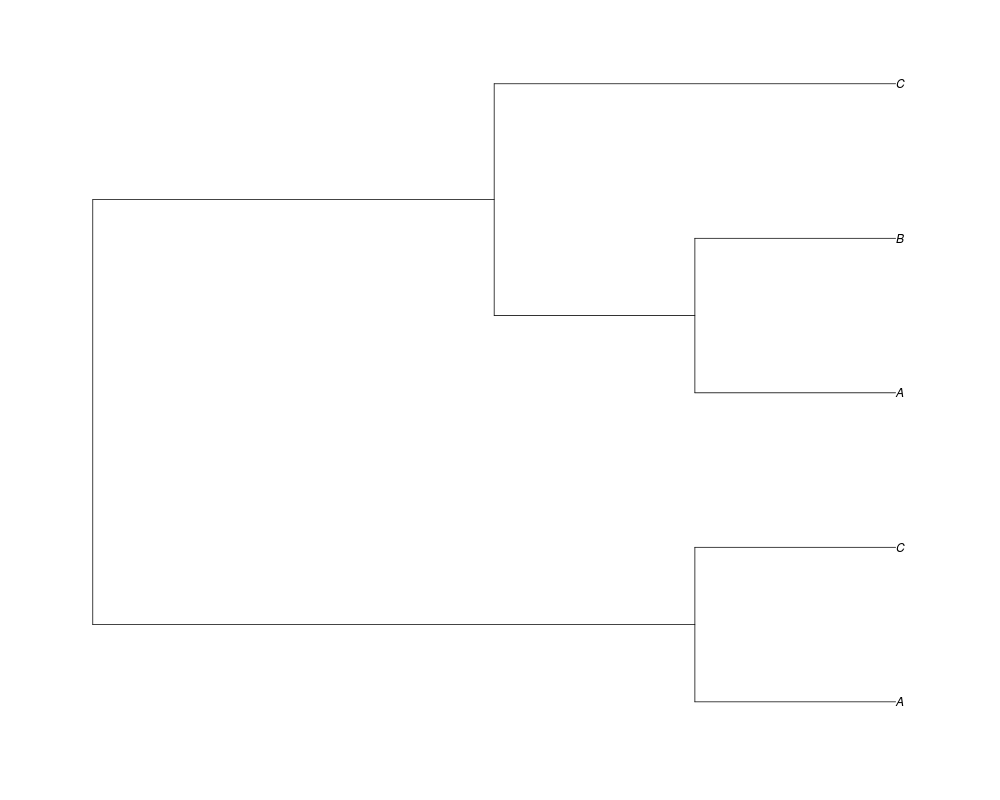
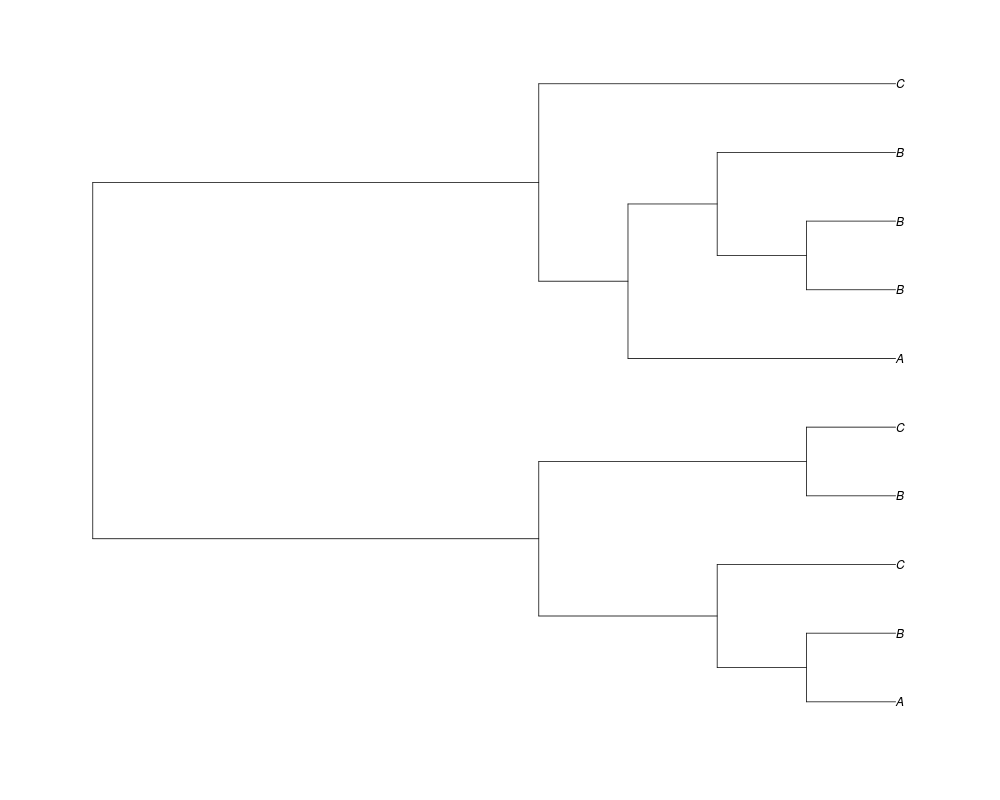
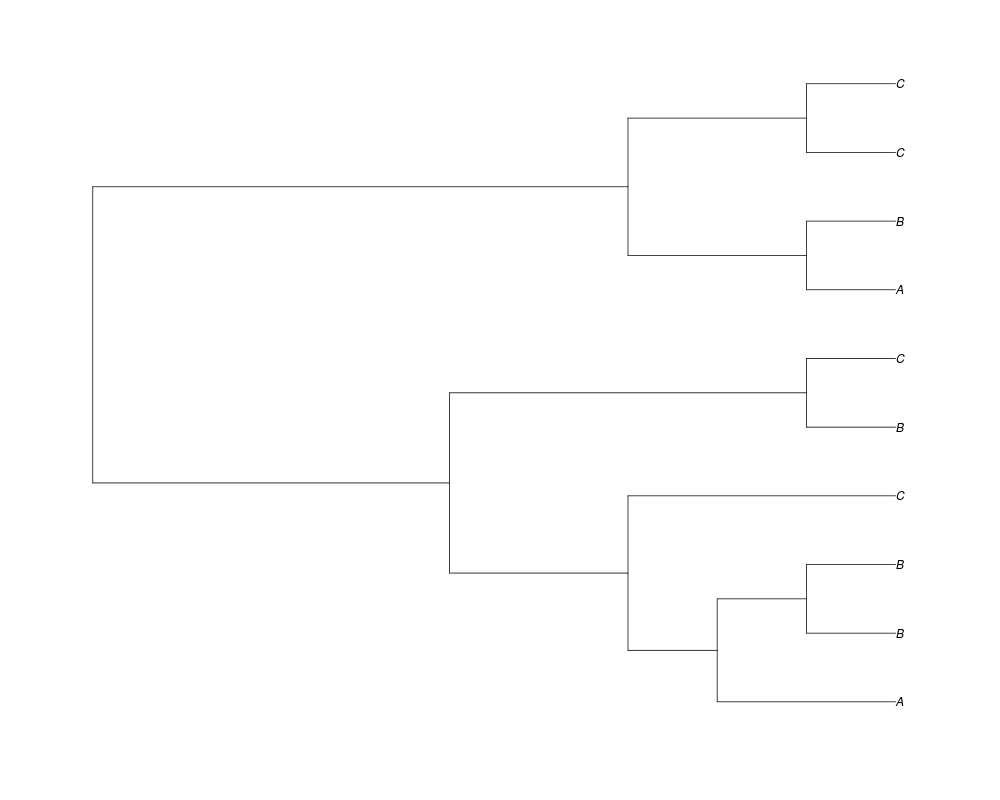
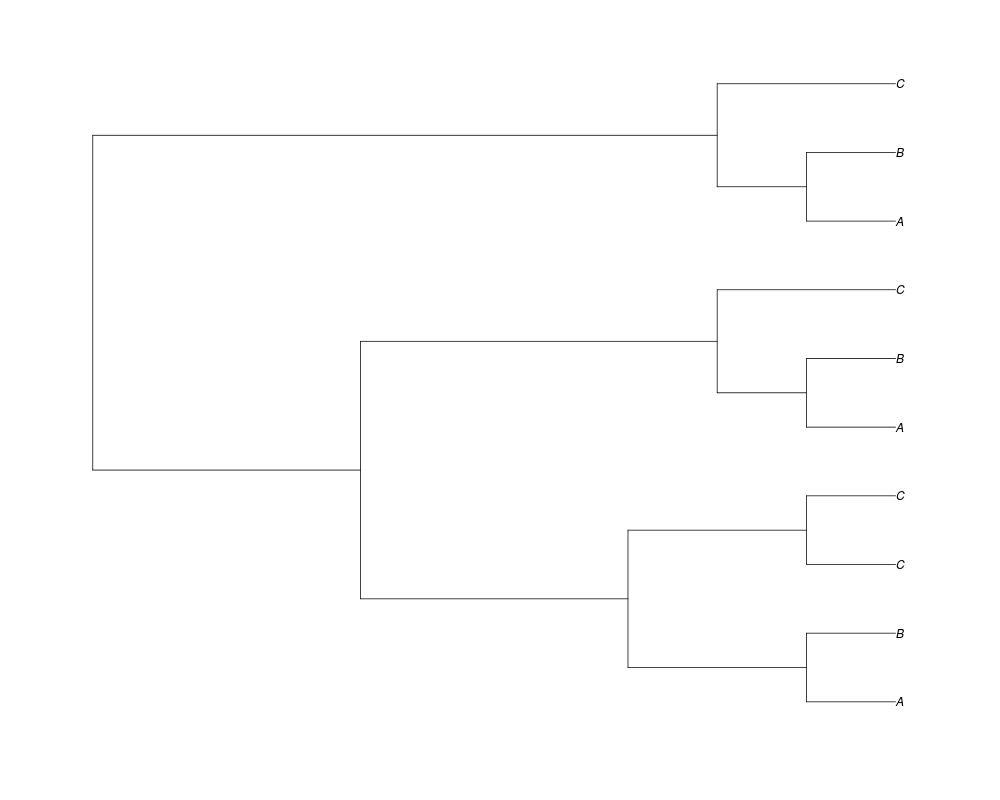
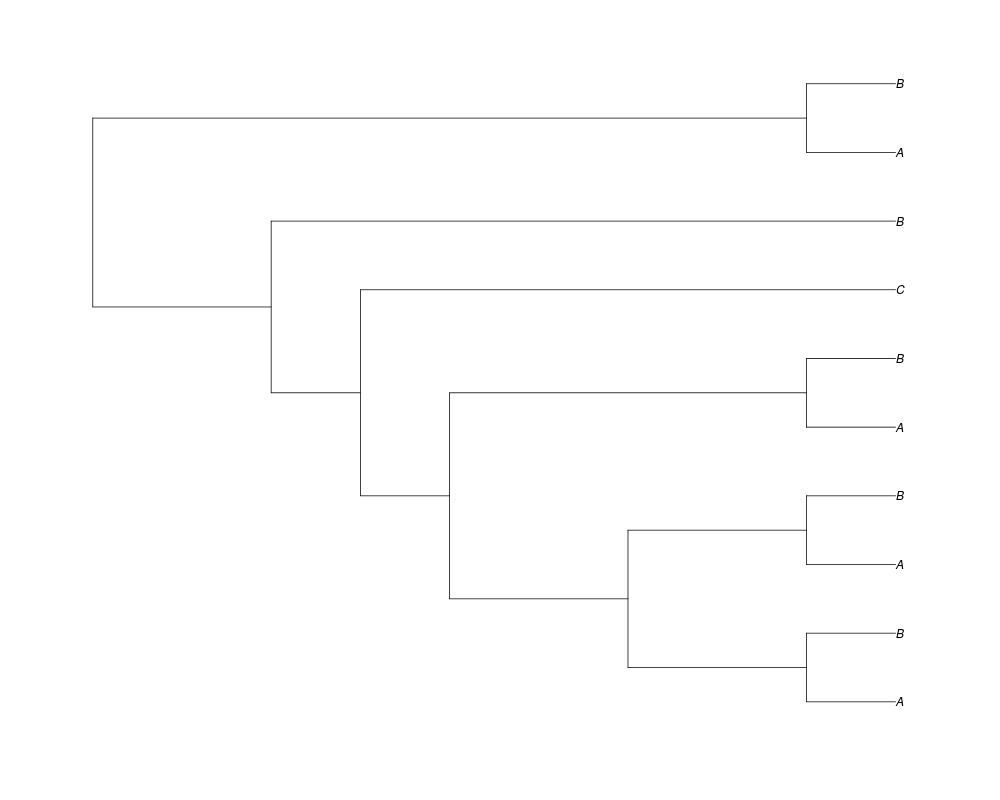
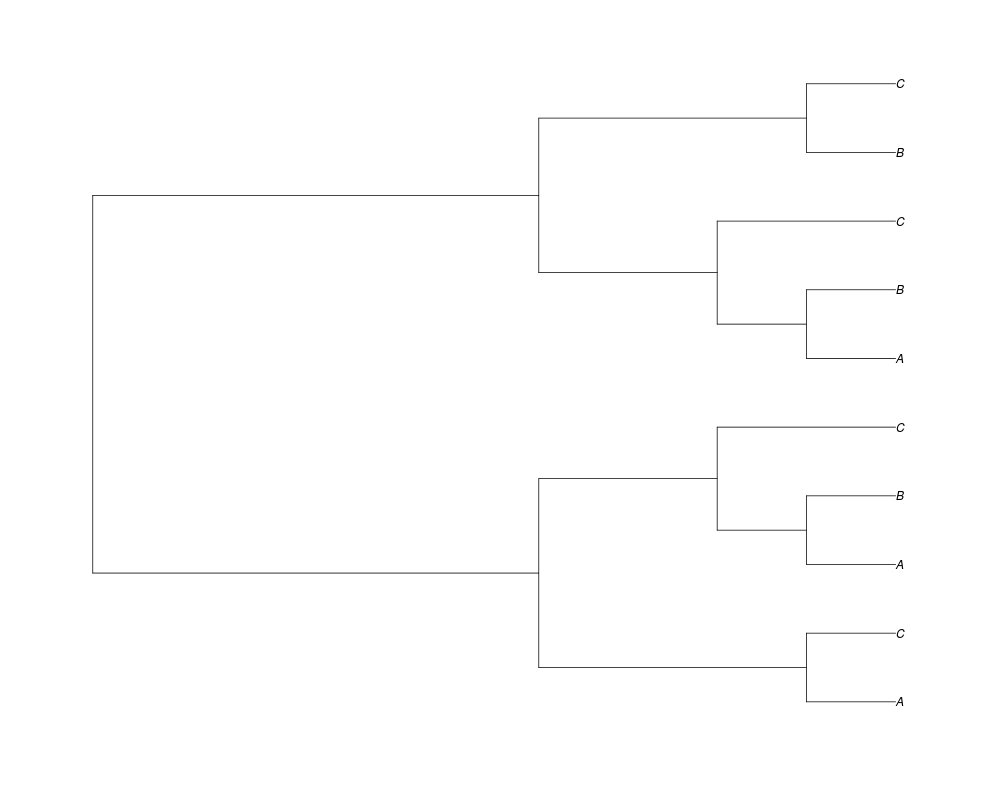
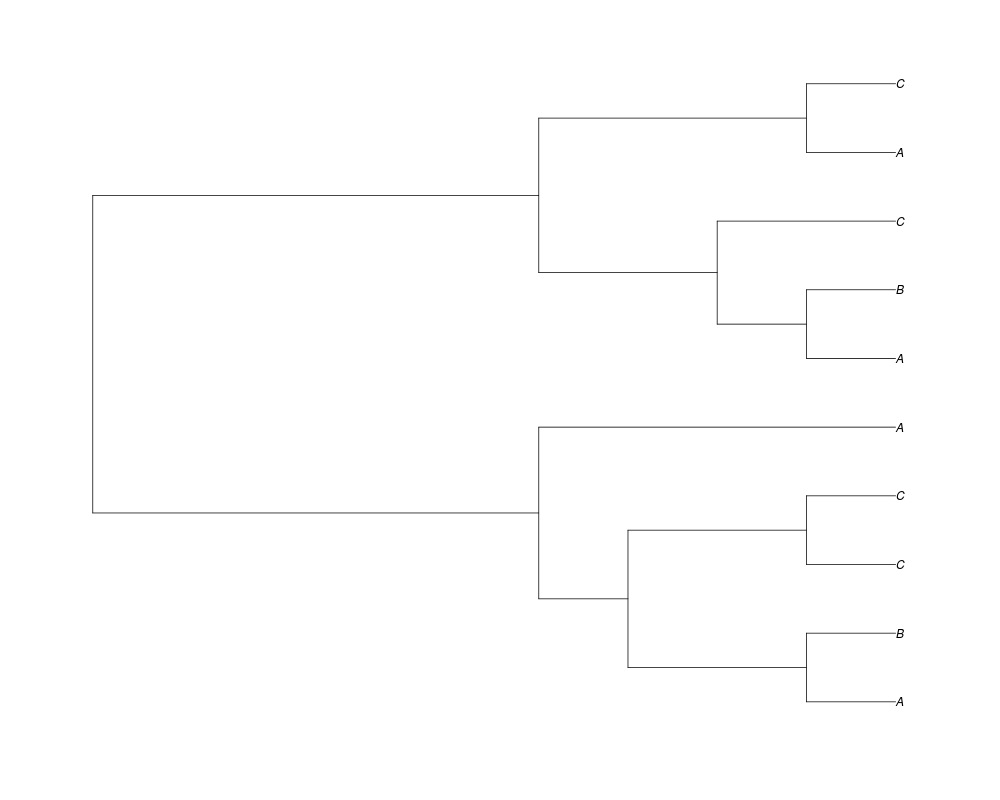
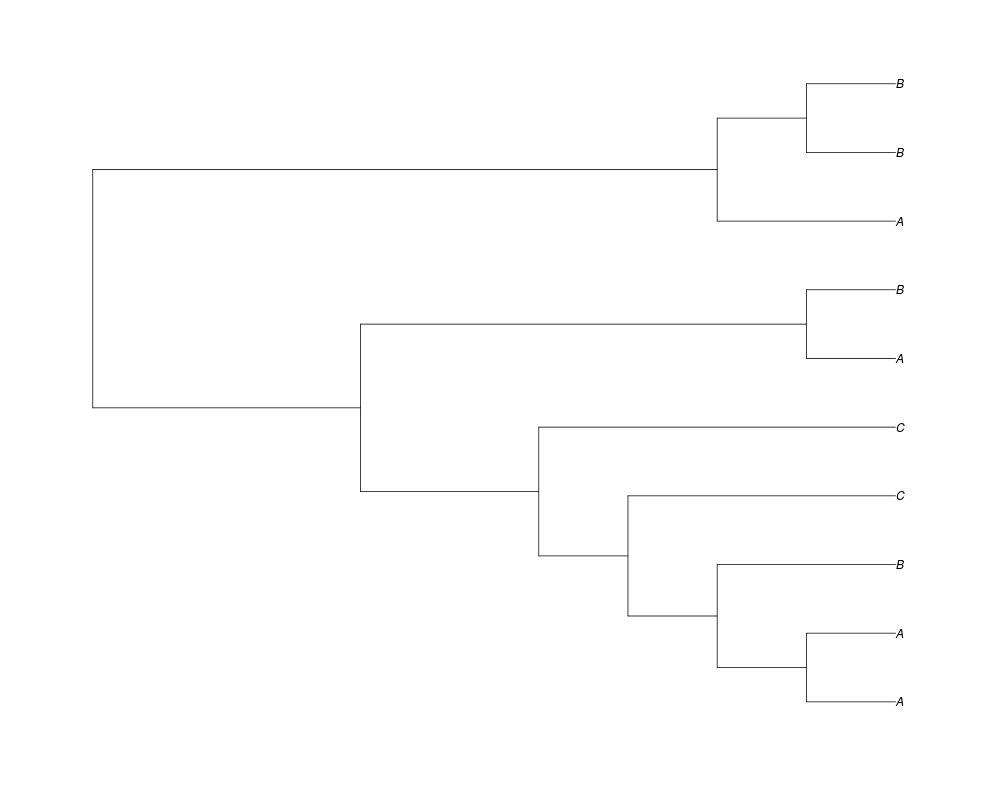
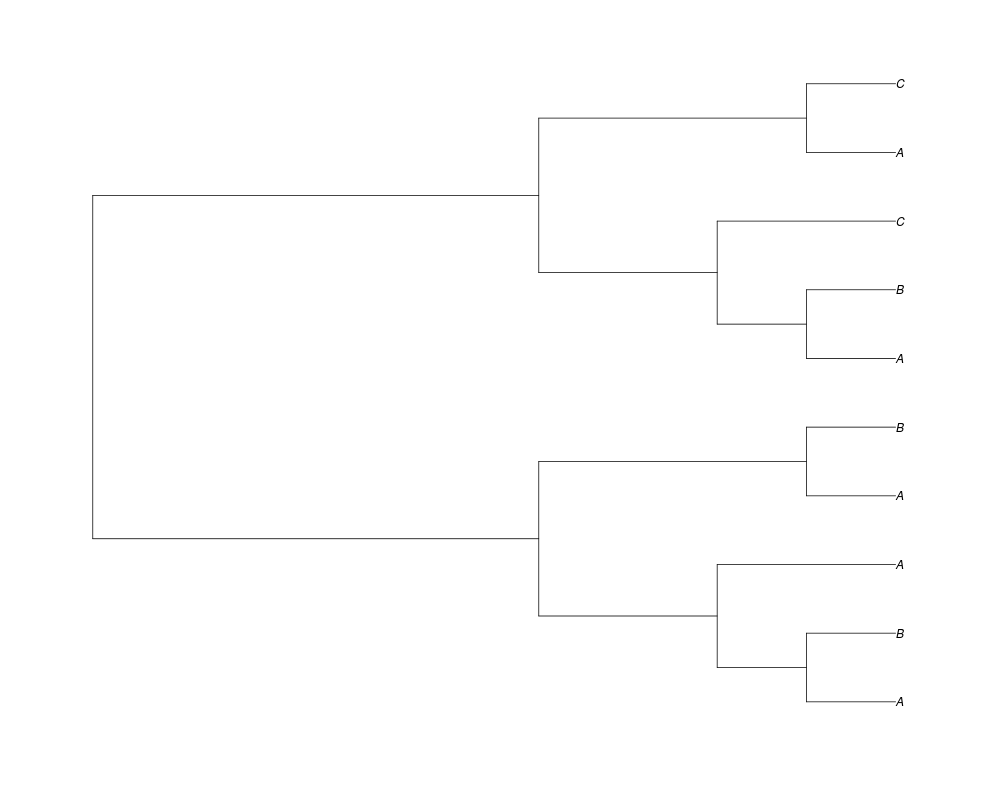
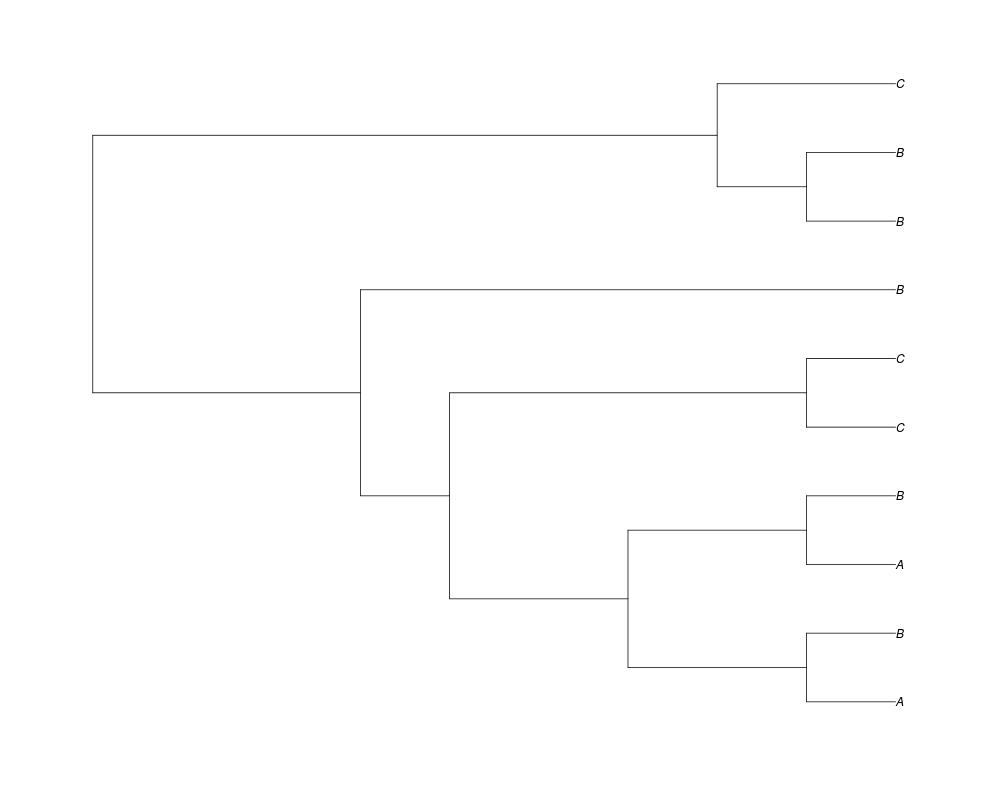
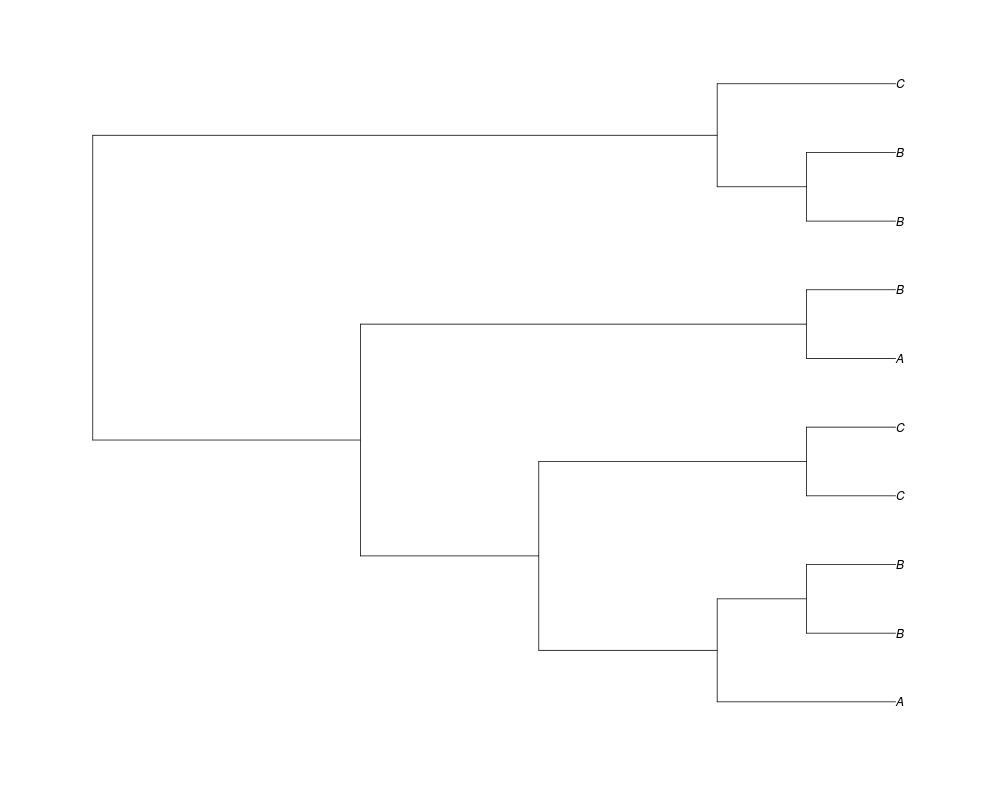
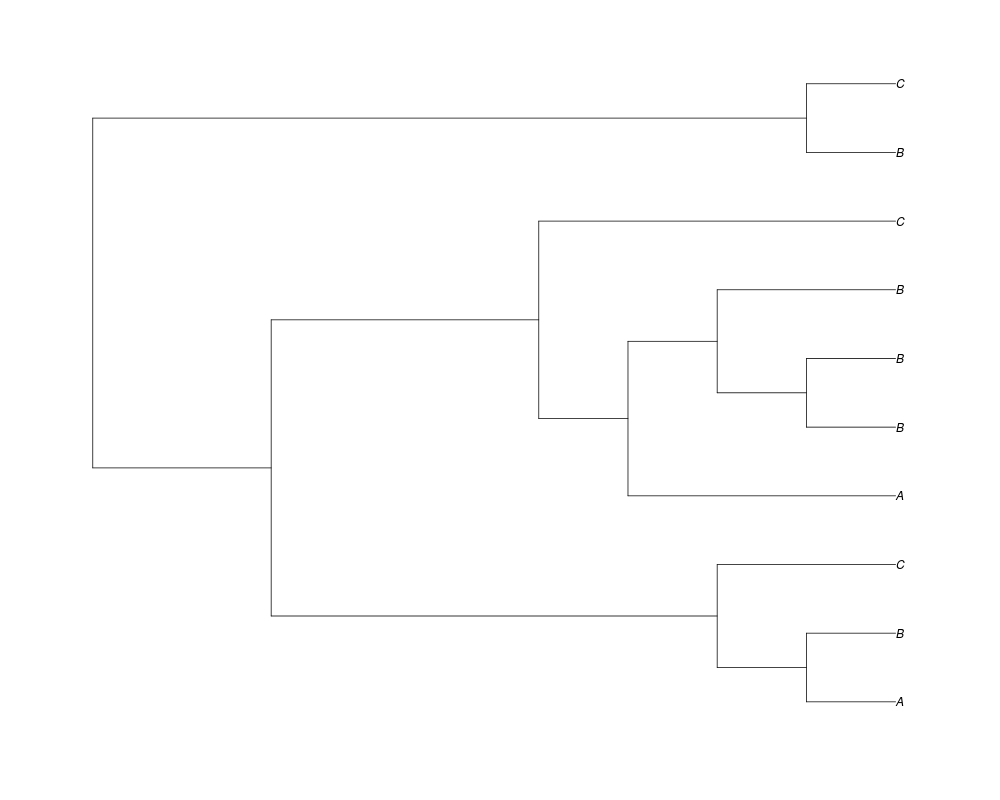
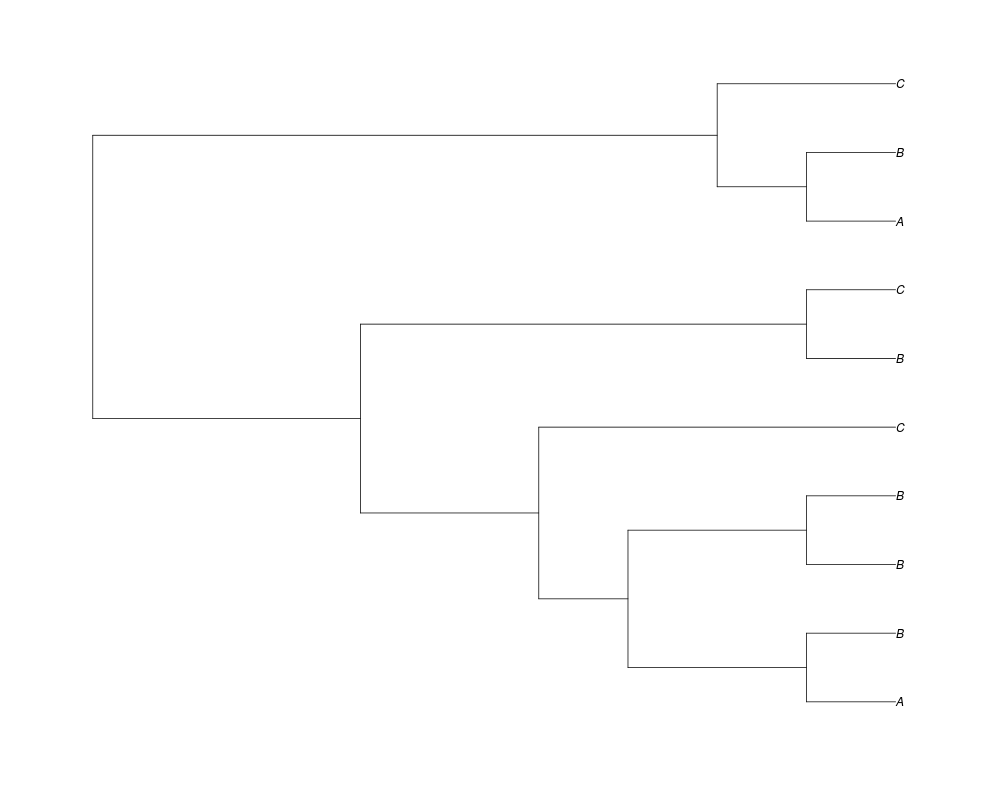
Examples##First we need a simple species tree spec<-read.tree(text="((A:0.5,B:0.5):0.5,C:1);") ##Now we sample ten gene trees starting with 3 reconstructed gene lineages phy.all<-rgenetree(10,spec,0.5,0.5,3) plot(phy.all) ##Now let's make sure that every tip has at least one gene and set an exponential prior on the root phy.full<-rgenetree(10,spec,0.5,0.5,exp(-(1:20))/sum(exp(-(1:20))),NULL,TRUE) plot(phy.full) ##Now lets force the whole gene tree to end in 10 genes and set a flat prior for the root phy.10<-rgenetree(10,spec,0.5,0.5,NULL,10) plot(phy.10) ##Now lets start with 3 genes, set the number of genes at each tip of spec and vary mu between the branches of spec phy.253<-rgenetree(10,spec,0.5,c(0,1,0.2,0.5),3,c(2,5,3)) plot(phy.253) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(HyPhy)
Loading required package: ape
Loading required package: R.utils
Loading required package: R.oo
Loading required package: R.methodsS3
R.methodsS3 v1.7.1 (2016-02-15) successfully loaded. See ?R.methodsS3 for help.
R.oo v1.20.0 (2016-02-17) successfully loaded. See ?R.oo for help.
Attaching package: 'R.oo'
The following objects are masked from 'package:methods':
getClasses, getMethods
The following objects are masked from 'package:base':
attach, detach, gc, load, save
R.utils v2.3.0 (2016-04-13) successfully loaded. See ?R.utils for help.
Attaching package: 'R.utils'
The following object is masked from 'package:utils':
timestamp
The following objects are masked from 'package:base':
cat, commandArgs, getOption, inherits, isOpen, parse, warnings
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/HyPhy/rgenetree.Rd_%03d_medium.png", width=480, height=480)
> ### Name: rgenetree
> ### Title: Samples gene family trees that evolved on a species tree with
> ### various conditions.
> ### Aliases: rgenetree
>
> ### ** Examples
>
> ##First we need a simple species tree
> spec<-read.tree(text="((A:0.5,B:0.5):0.5,C:1);")
> ##Now we sample ten gene trees starting with 3 reconstructed gene lineages
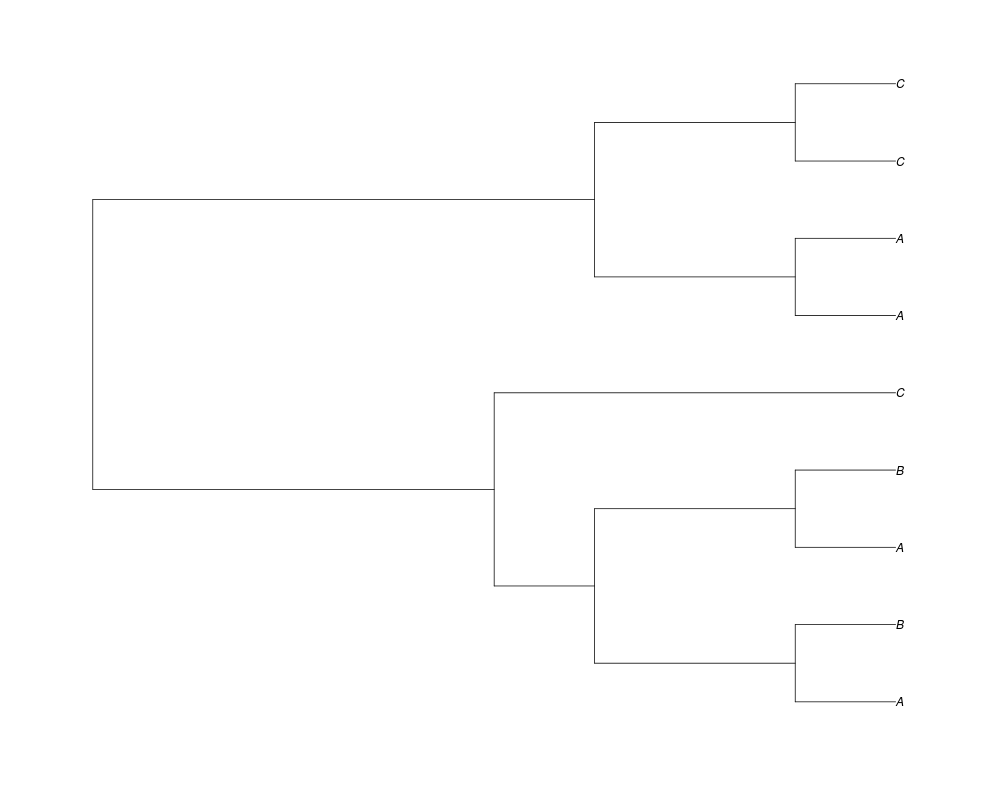
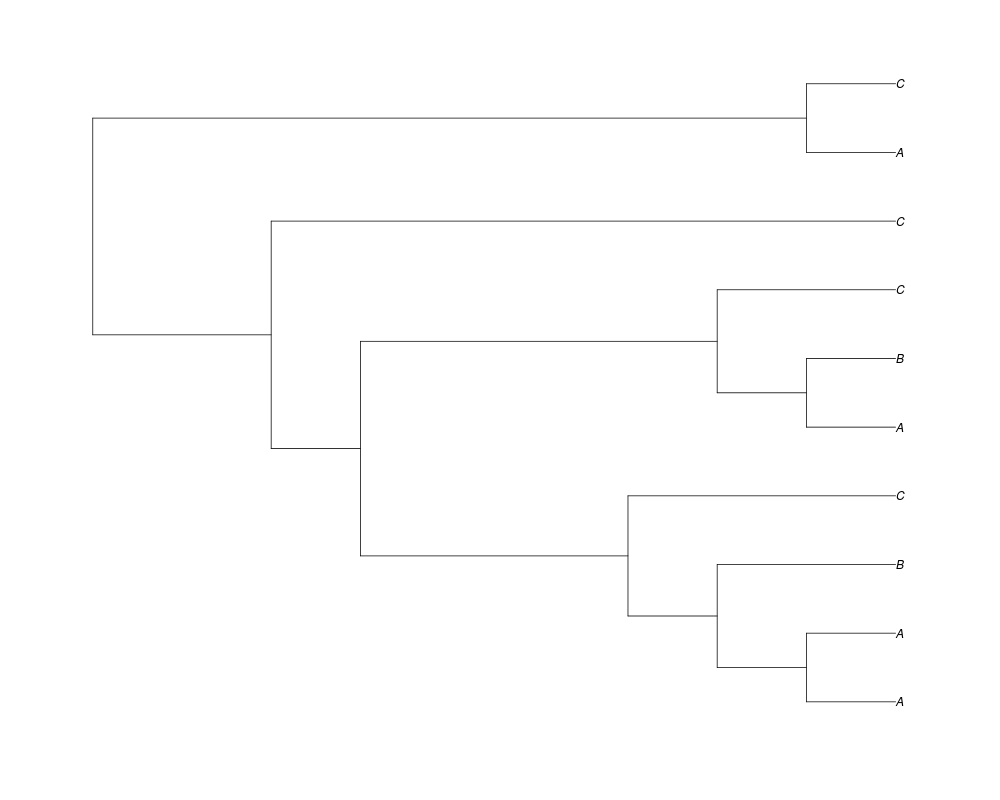
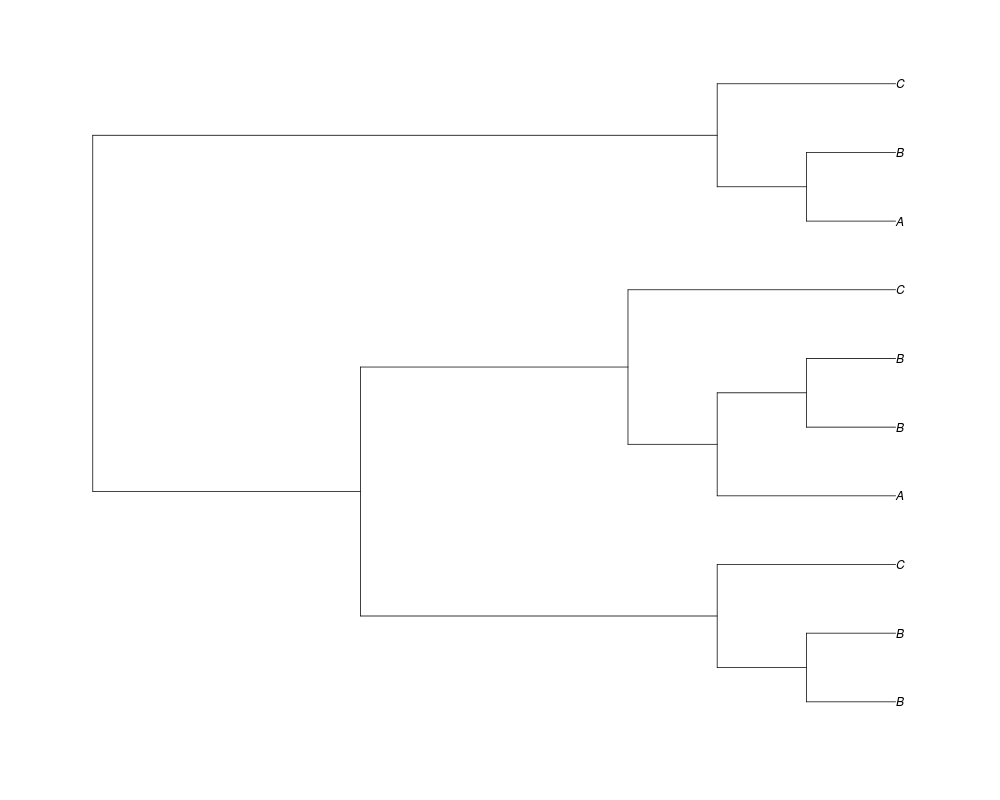
> phy.all<-rgenetree(10,spec,0.5,0.5,3)
> plot(phy.all)
>
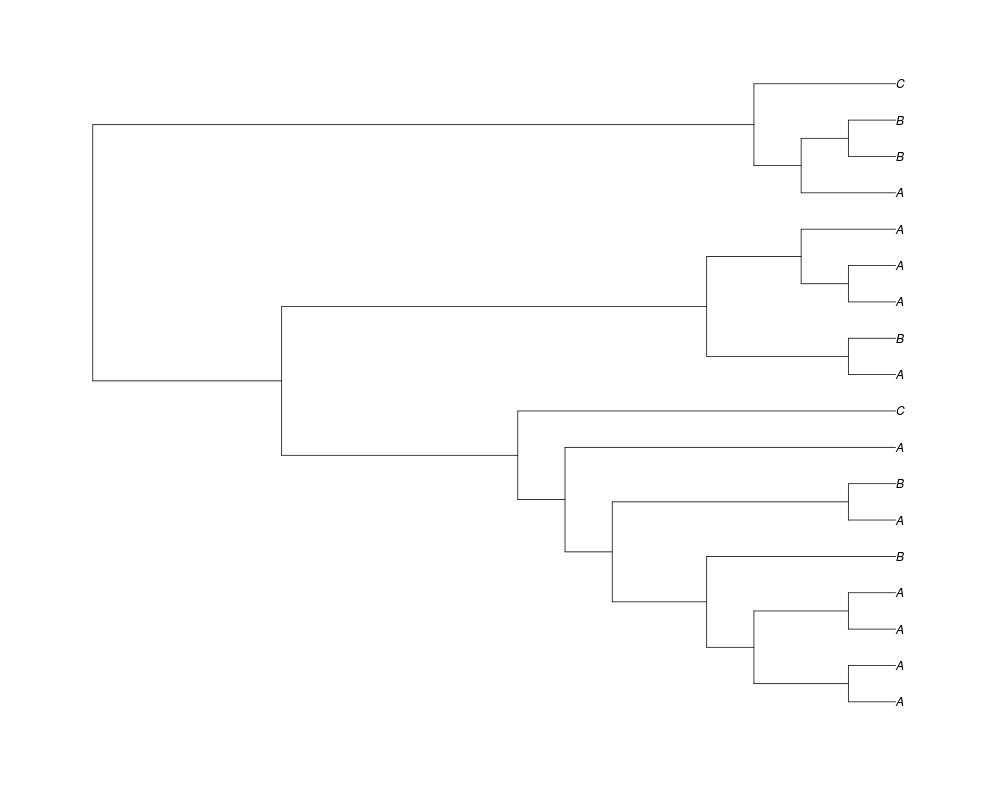
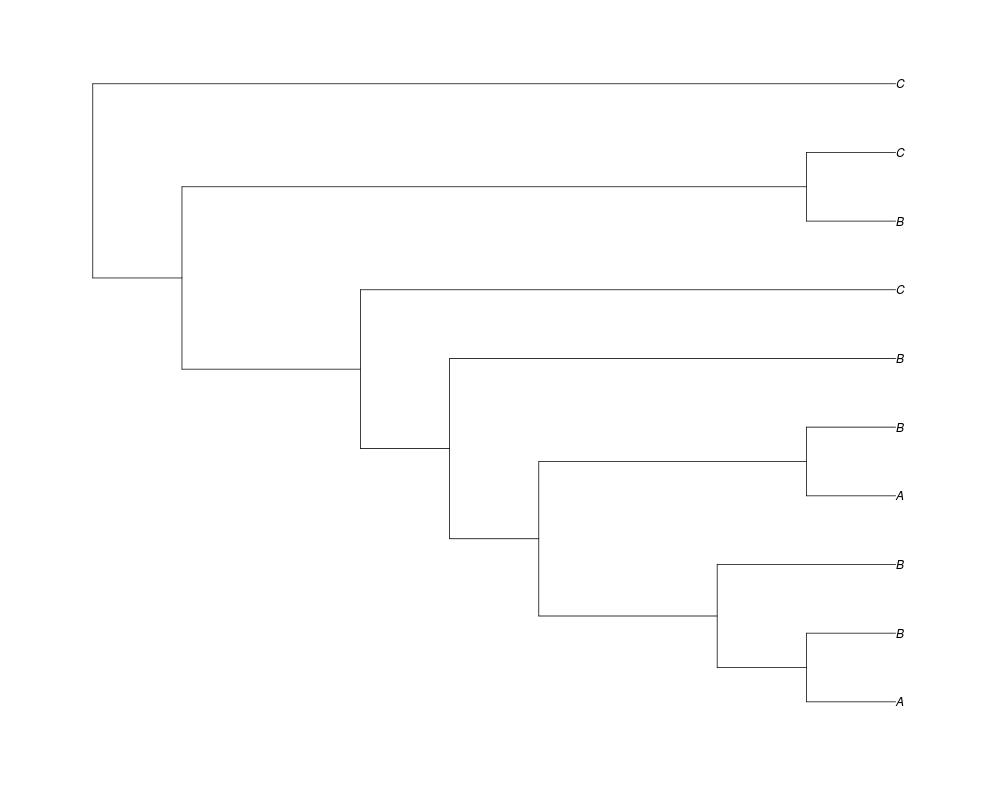
> ##Now let's make sure that every tip has at least one gene and set an exponential prior on the root
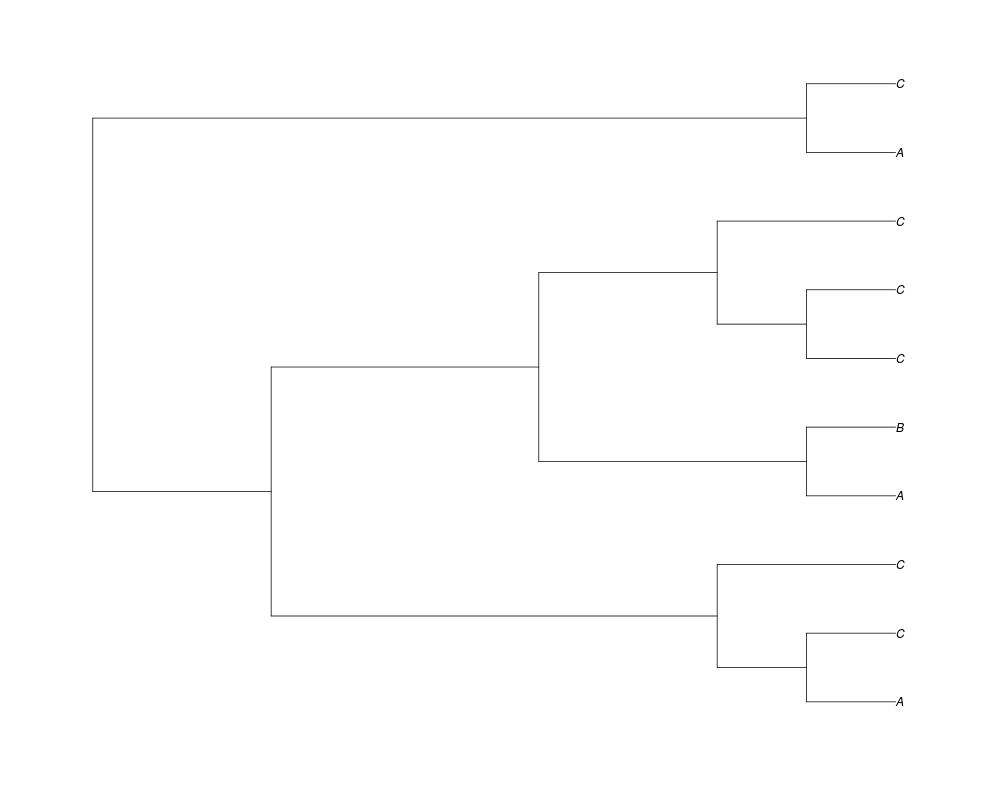
> phy.full<-rgenetree(10,spec,0.5,0.5,exp(-(1:20))/sum(exp(-(1:20))),NULL,TRUE)
> plot(phy.full)
>
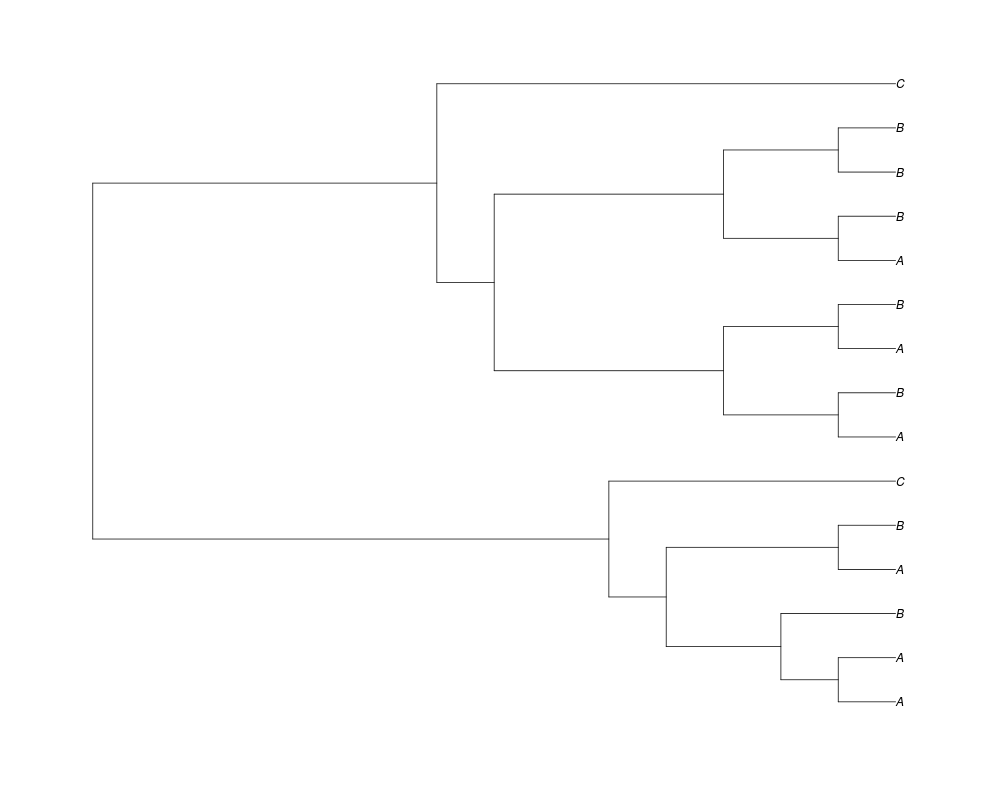
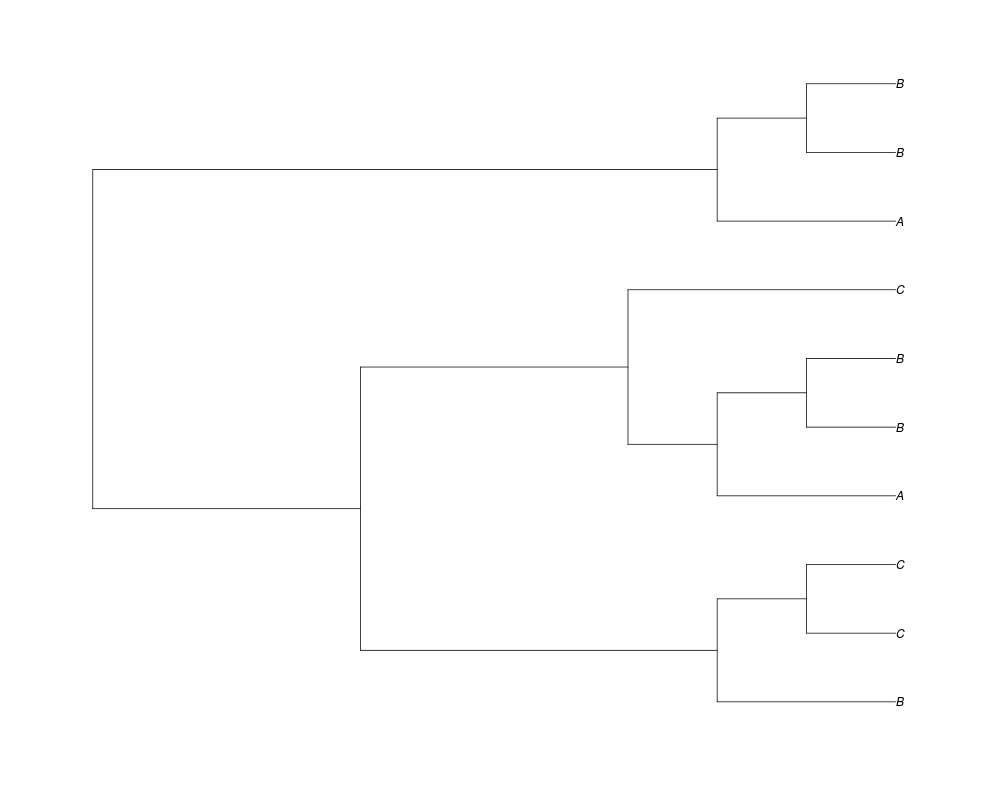
> ##Now lets force the whole gene tree to end in 10 genes and set a flat prior for the root
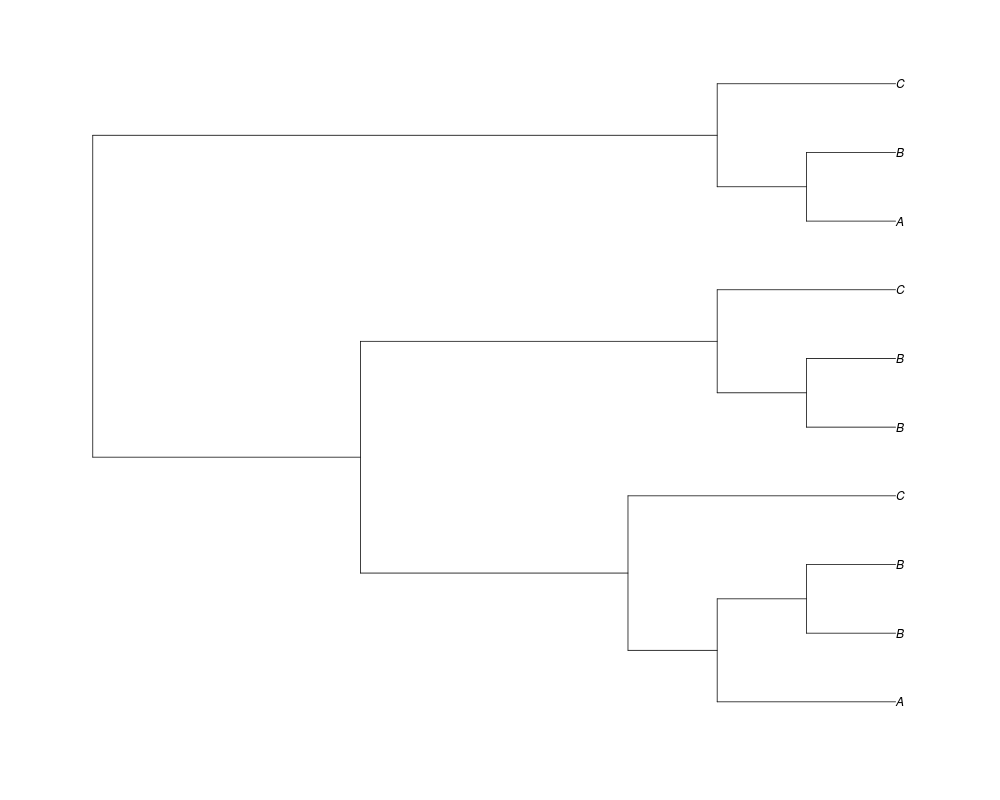
> phy.10<-rgenetree(10,spec,0.5,0.5,NULL,10)
> plot(phy.10)
>
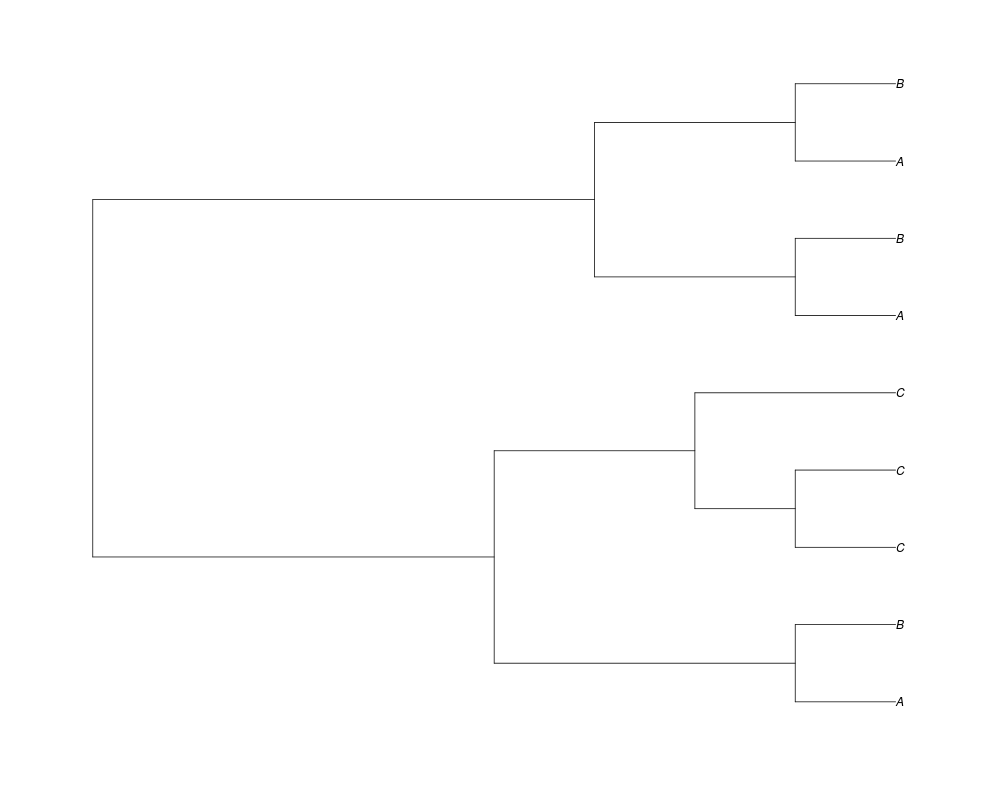
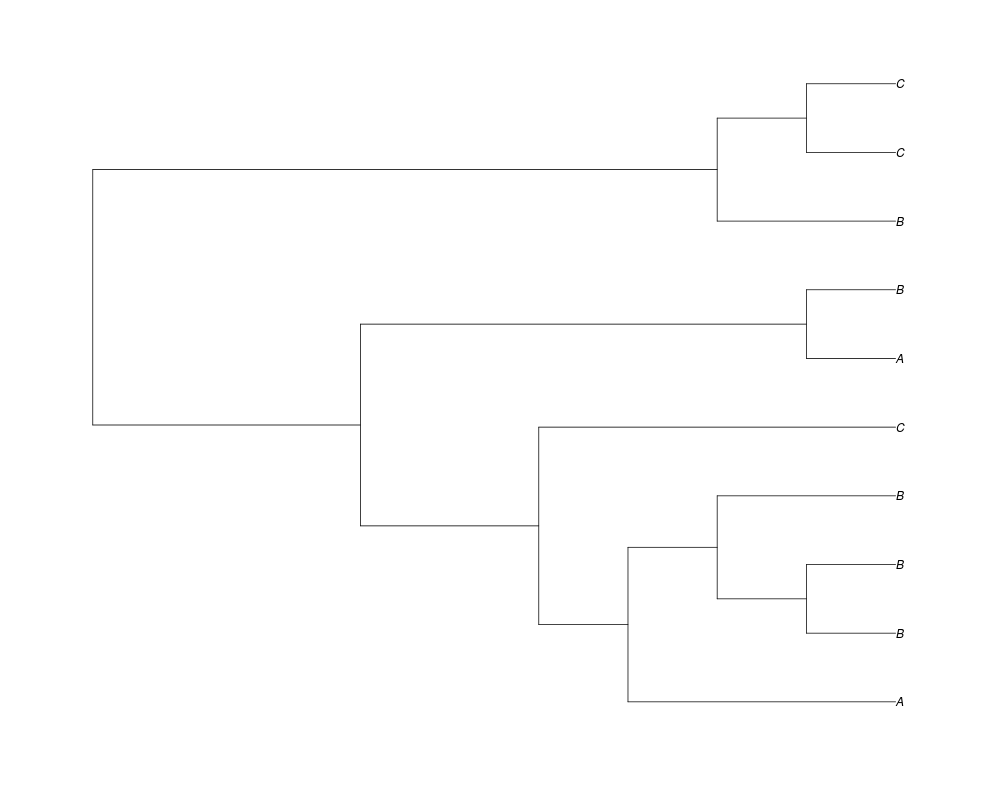
> ##Now lets start with 3 genes, set the number of genes at each tip of spec and vary mu between the branches of spec
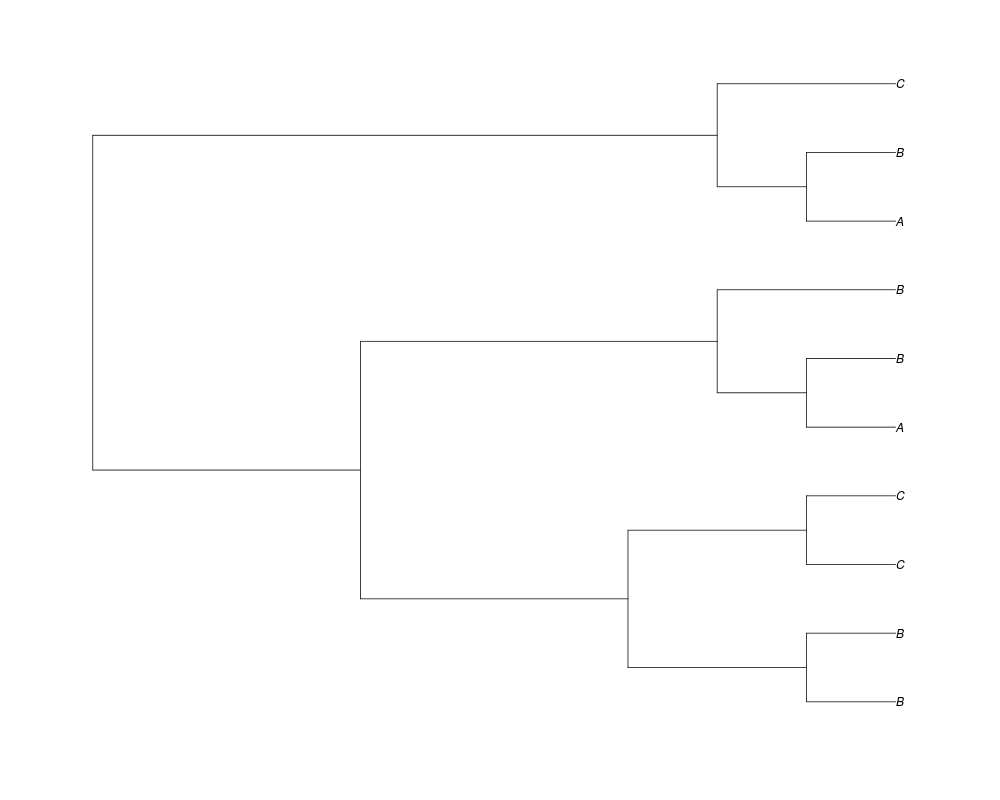
> phy.253<-rgenetree(10,spec,0.5,c(0,1,0.2,0.5),3,c(2,5,3))
> plot(phy.253)
>
>
>
>
>
> dev.off()
null device
1
>
|