Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
IBD simulationDescriptionThis is the main function of the package. Gene dropping of chromosomes is simulated down the pedigree, either unconditionally or conditional on the 'condition' pattern if this is given. Regions showing the 'query' pattern are collected and summarized. Usage
IBDsim(x, sims, query=NULL, condition=NULL, map="decode",
chromosomes=NULL, model="chi", merged=TRUE, simdata=NULL,
skip.recomb = "noninf_founders", seed=NULL, verbose=TRUE)
Arguments
DetailsEach simulation starts by unique alleles being distributed to the pedigree founders. In each subsequent meiosis, homologue chromosomes are made to recombine according to a renewal process along the four-strand bundle, with chi square distributed waiting times. (For comparison purposes, Haldane's Poisson model for recombination is also implemented.) Recombination rates are sex-dependent, and vary along each chromosome according to the recombination map specified by the IBD patterns are described as combinations of Single Allele Patterns (SAPs). A SAP is a specification for a given allele of the number of copies carried by various individuals, and must be given as a list of numerical vectors (containing ID labels) named '0', '1', '2', 'atleast1' and 'atmost1' (some of these can be absent or NULL; see Examples). If a condition SAP is given (i.e. if ValueIf the If
The term 'IBD segment' in the above always refers to 'IBD segment matching the query SAP'.
The suffixes Author(s)Magnus Dehli Vigeland ReferencesThe Decode map: Kong, A. et al. (2010) Fine scale recombination rate differences between sexes, populations and individuals. Nature, 467, 1099–1103. doi:10.1038/nature09525. The chi-square model: Zhao, H., Speed, T. P., McPeek, M. S. (1995) Statistical analysis of crossover interference using the chi-square model. Genetics, 139(2), 1045–1056. Examples
# In all examples below, the 'sims' parameter is set to 1 to
# decrease runtime. For realistic results increase to e.g. 1000.
#### An example with a recessive disease in a consanguineous pedigree:
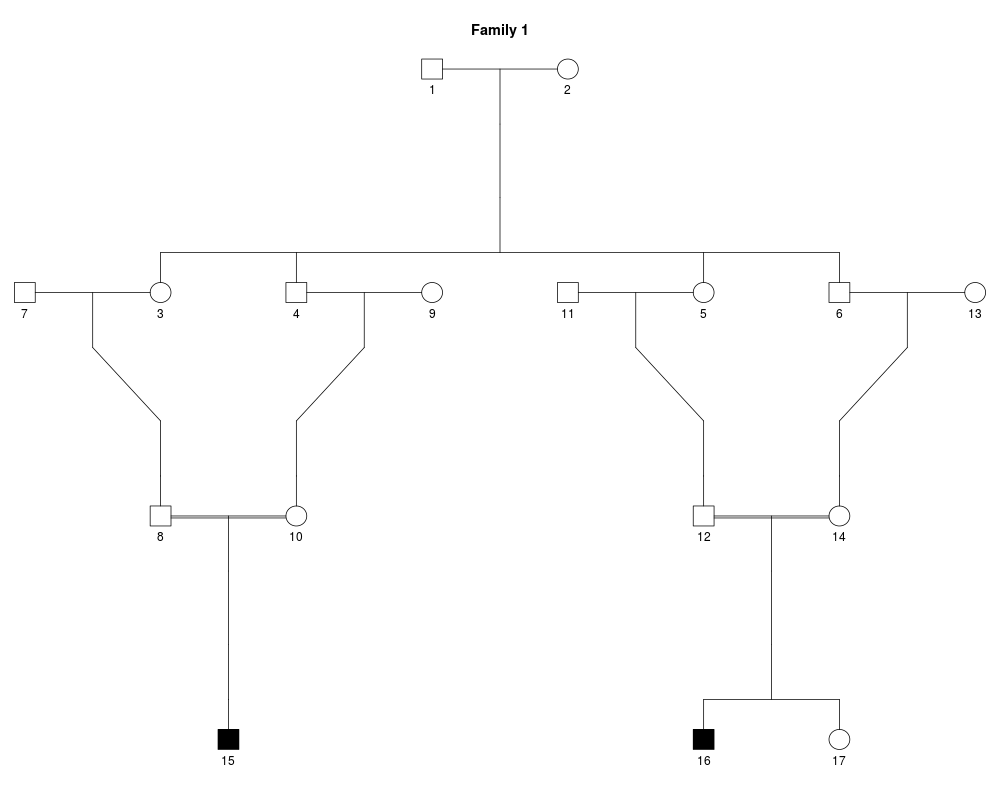
x = linkdat(twoloops)
plot(x)
# If individuals 15, 16 and 17 are available for sequencing, we can
# predict the number and size of disease-compatible IBD segments as
# follows:
sap1 = list('2'=15:16, 'atmost1'=17)
IBDsim(x, sims=1, condition=sap1, query=sap1)
# If 16 is unavailable, his parents and healthy sibling are still
# informative. The regions we are looking for now are those with
# an allele present in 2 copies in 15, 1 copy in 12 and 14, and
# at most one in 17. Note that the condition SAP remains as above.
IBDsim(x, sims=1, condition=sap1,
query=list('2'=15, '1'=c(12,14), 'atmost1'=17))
#### Example with an autosomal dominant disorder:
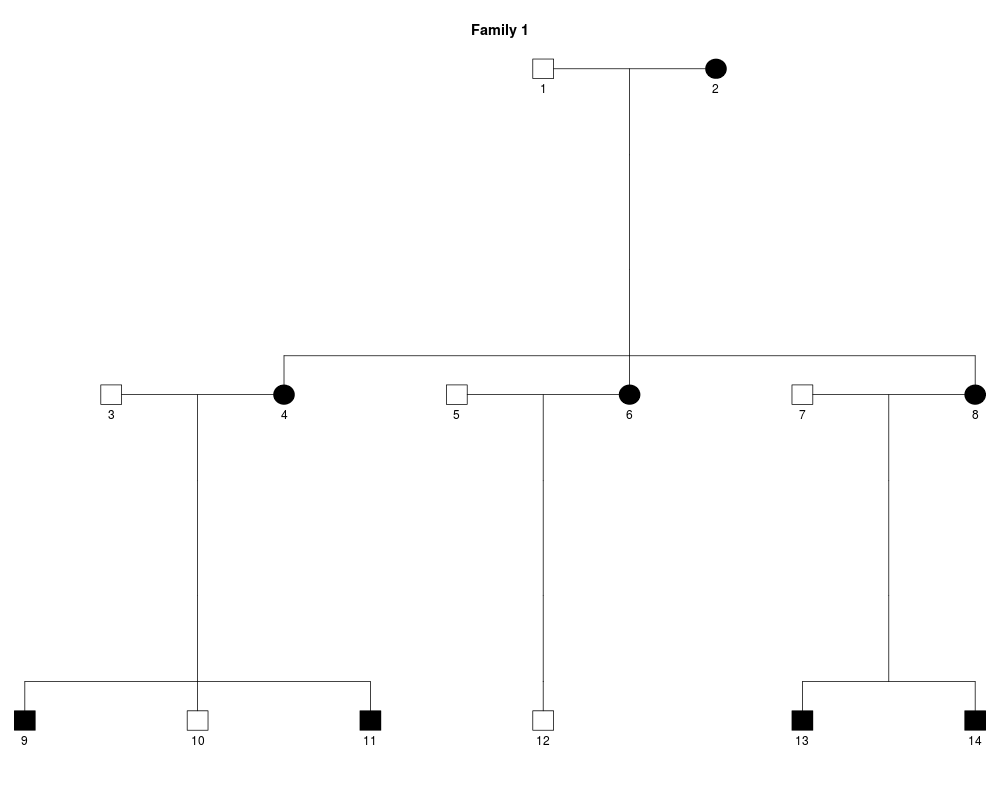
y = linkdat(dominant1) # lazy load data
plot(y)
# Suppose a 20 Mb linkage peak is found.
# How often would this occur by chance?
aff = which(dominant1[,'AFF']==2) # the affected
nonaff = which(dominant1[,'AFF']==1) # the non-affected
dom_pattern = list('1'=aff, '0'=nonaff)
res = IBDsim(y, sims=1, query=dom_pattern)
mean(res$stats['longest.all',] > 20) # the estimated p-value
#### Another example: Unconditional simulation of regions shared
# IBD by third cousins. The "zeroprob" entry in the output shows
# the percentage of simulations having no IBD-sharing among the
# two cousins. (Again: Increase 'sims' for more accurate results.)
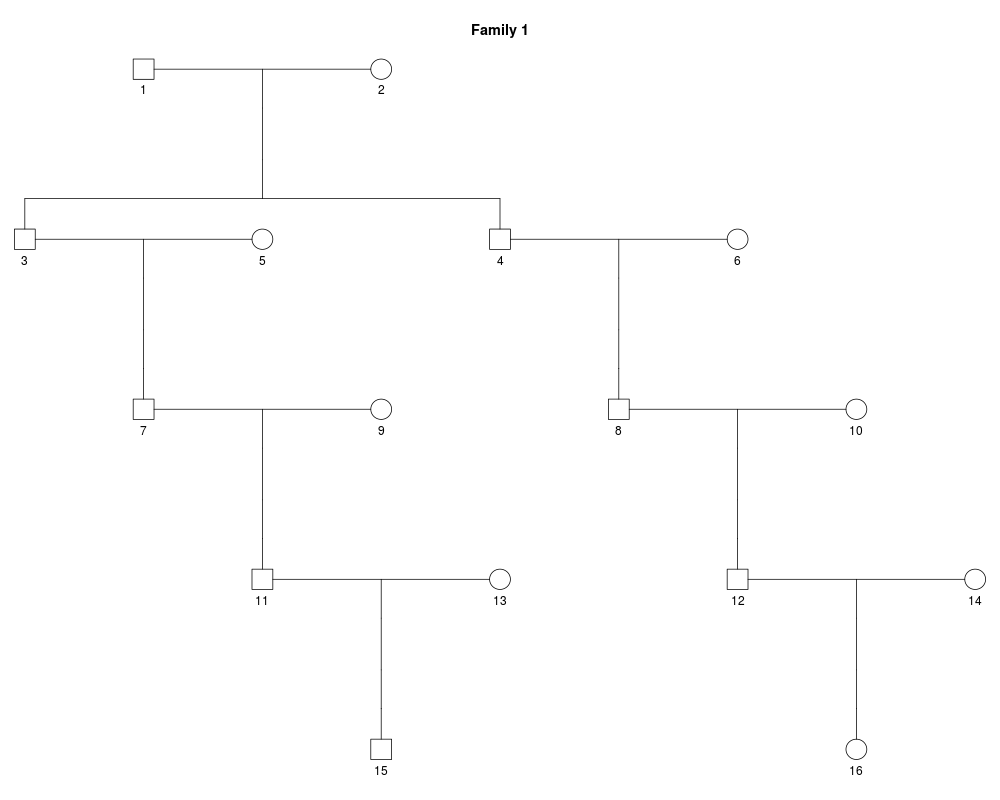
x_male = cousinPed(3)
plot(x_male)
IBDsim(x_male, sims=1, query=list('1'=15:16))
# Changing the genders of the individuals connecting the cousins
# can have a big impact on the distribution of IBD segments:
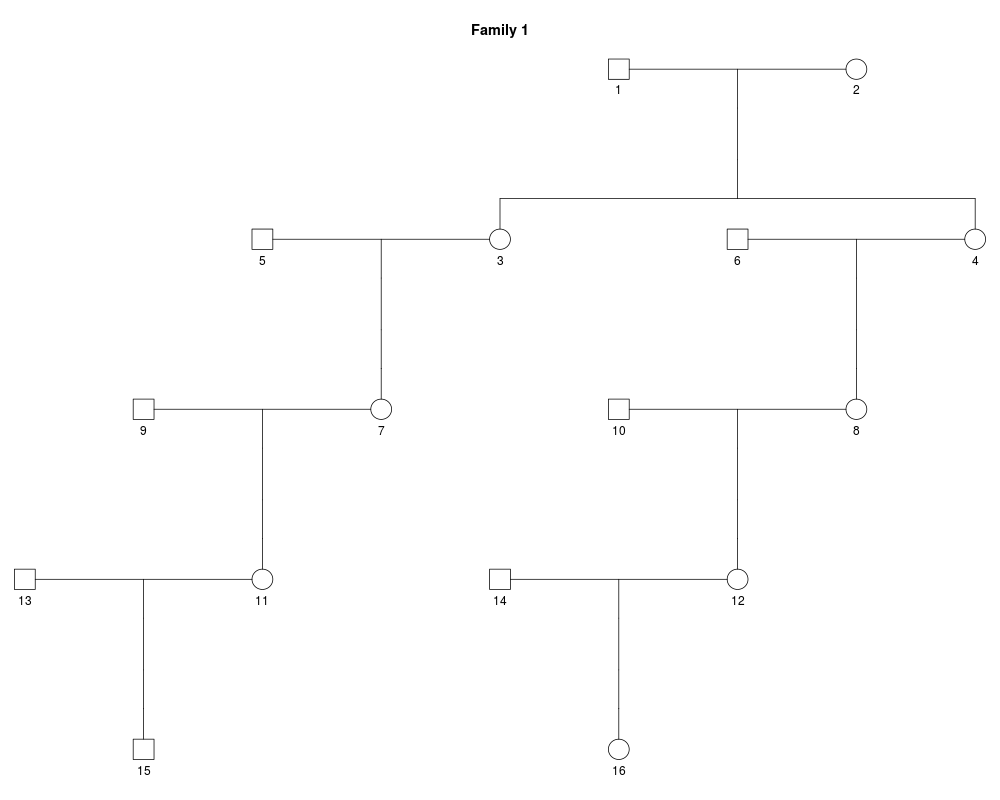
x_female = swapSex(x_male, c(3,4,7,8,11,12))
plot(x_female)
IBDsim(x_female, sims=1, query=list('1'=15:16))
## Given that the two third cousins have at least one segment in
# common, what is the expected length of this segment? We simulate
# conditional on one allele in common between the cousins. The
# "length.dis" entry of the summary shows the average length of
# the disease region (which should be quite a lot larger than
# an average random segment).
sap = list('1'=15:16)
IBDsim(x_male, sims=1, condition=sap, query=sap)
#### Let us look at a different relationship: Half first cousins.
# Two such cousins will on average share 1/8 = 12.5% of the autosome.
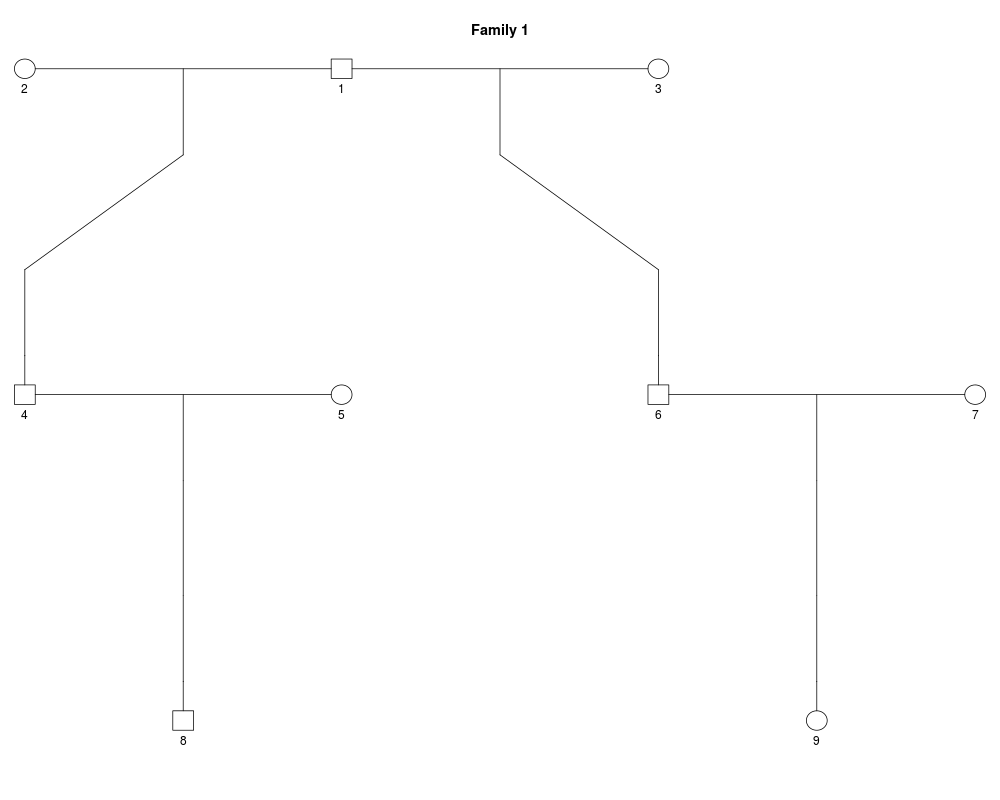
z = halfCousinPed(1)
plot(z)
res = IBDsim(z, sims=1, query=list('1'=8:9))
res$stats
# visualizing the spread in total IBD sharing and the number of segments:
IBD_percent = res$stats['fraction.all', ]
IBD_count = res$stats['count.all', ]
hist(IBD_percent)
hist(IBD_count)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(IBDsim)
Loading required package: paramlink
Loading required package: kinship2
Loading required package: Matrix
Loading required package: quadprog
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/IBDsim/IBDsim.Rd_%03d_medium.png", width=480, height=480)
> ### Name: IBDsim
> ### Title: IBD simulation
> ### Aliases: IBDsim
> ### Keywords: math
>
> ### ** Examples
>
> # In all examples below, the 'sims' parameter is set to 1 to
> # decrease runtime. For realistic results increase to e.g. 1000.
>
> #### An example with a recessive disease in a consanguineous pedigree:
>
> x = linkdat(twoloops)
Family ID: 1.
17 individuals.
2 affected 15 non-affected.
Loop(s) detected.
0 markers.
> plot(x)
>
> # If individuals 15, 16 and 17 are available for sequencing, we can
> # predict the number and size of disease-compatible IBD segments as
> # follows:
>
> sap1 = list('2'=15:16, 'atmost1'=17)
> IBDsim(x, sims=1, condition=sap1, query=sap1)
---------------
Performing conditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
For the disease chromosome I'm sampling condition SAPs among the following:
SAP 1:
Two copies: 15, 16
One copy: 1, 3, 4, 5, 6, 8, 10, 12, 14
At most one copy: 17
SAP 2:
Two copies: 15, 16
One copy: 2, 3, 4, 5, 6, 8, 10, 12, 14
At most one copy: 17
Skipping recombination in the following individuals: 7, 9, 11, 13
Simulation finished. Time used: 0.274 seconds
---------------
Inbreeding coefficients:
ID f
15 0.0625
16 0.0625
17 0.0625
Results:
count.all fraction.all average.all longest.all count.rand
2.000 0.965 13.823 25.805 1.000
fraction.rand average.rand longest.rand length.dis rank.dis
0.064 1.840 1.840 25.805 1.000
zeroprob
0.000
Total time used: 0.303 seconds.
>
> # If 16 is unavailable, his parents and healthy sibling are still
> # informative. The regions we are looking for now are those with
> # an allele present in 2 copies in 15, 1 copy in 12 and 14, and
> # at most one in 17. Note that the condition SAP remains as above.
>
> IBDsim(x, sims=1, condition=sap1,
+ query=list('2'=15, '1'=c(12,14), 'atmost1'=17))
---------------
Performing conditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
For the disease chromosome I'm sampling condition SAPs among the following:
SAP 1:
Two copies: 15, 16
One copy: 1, 3, 4, 5, 6, 8, 10, 12, 14
At most one copy: 17
SAP 2:
Two copies: 15, 16
One copy: 2, 3, 4, 5, 6, 8, 10, 12, 14
At most one copy: 17
Skipping recombination in the following individuals: 7, 9, 11, 13
Simulation finished. Time used: 0.122 seconds
---------------
Inbreeding coefficients:
ID f
15 0.0625
16 0.0625
17 0.0625
Results:
count.all fraction.all average.all longest.all count.rand
2.000 0.251 3.595 4.503 1.000
fraction.rand average.rand longest.rand length.dis rank.dis
0.094 2.687 2.687 4.503 1.000
zeroprob
0.000
Total time used: 0.148 seconds.
>
>
> #### Example with an autosomal dominant disorder:
> y = linkdat(dominant1) # lazy load data
Family ID: 1.
14 individuals.
8 affected 6 non-affected.
4 nuclear subfamilies.
0 markers.
> plot(y)
>
> # Suppose a 20 Mb linkage peak is found.
> # How often would this occur by chance?
>
> aff = which(dominant1[,'AFF']==2) # the affected
> nonaff = which(dominant1[,'AFF']==1) # the non-affected
> dom_pattern = list('1'=aff, '0'=nonaff)
> res = IBDsim(y, sims=1, query=dom_pattern)
---------------
Performing unconditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
Skipping recombination in the following individuals: 1, 3, 5, 7
Simulation finished. Time used: 0.064 seconds
---------------
Results:
count.all fraction.all average.all longest.all zeroprob
0 0 0 0 100
Total time used: 0.127 seconds.
> mean(res$stats['longest.all',] > 20) # the estimated p-value
[1] 0
>
>
> #### Another example: Unconditional simulation of regions shared
> # IBD by third cousins. The "zeroprob" entry in the output shows
> # the percentage of simulations having no IBD-sharing among the
> # two cousins. (Again: Increase 'sims' for more accurate results.)
>
> x_male = cousinPed(3)
> plot(x_male)
> IBDsim(x_male, sims=1, query=list('1'=15:16))
---------------
Performing unconditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
Skipping recombination in the following individuals: 5, 6, 9, 10, 13, 14
Simulation finished. Time used: 0.145 seconds
---------------
Results:
count.all fraction.all average.all longest.all zeroprob
2.000 1.027 14.715 20.898 0.000
Total time used: 0.154 seconds.
>
> # Changing the genders of the individuals connecting the cousins
> # can have a big impact on the distribution of IBD segments:
>
> x_female = swapSex(x_male, c(3,4,7,8,11,12))
Changing sex of the following spouses as well: 5, 6, 9, 10, 13, 14
> plot(x_female)
> IBDsim(x_female, sims=1, query=list('1'=15:16))
---------------
Performing unconditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
Skipping recombination in the following individuals: 5, 6, 9, 10, 13, 14
Simulation finished. Time used: 0.061 seconds
---------------
Results:
count.all fraction.all average.all longest.all zeroprob
5.000 0.751 4.301 15.273 0.000
Total time used: 0.076 seconds.
>
> ## Given that the two third cousins have at least one segment in
> # common, what is the expected length of this segment? We simulate
> # conditional on one allele in common between the cousins. The
> # "length.dis" entry of the summary shows the average length of
> # the disease region (which should be quite a lot larger than
> # an average random segment).
>
> sap = list('1'=15:16)
> IBDsim(x_male, sims=1, condition=sap, query=sap)
---------------
Performing conditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
For the disease chromosome I'm sampling condition SAPs among the following:
SAP 1:
One copy: 1, 3, 4, 7, 8, 11, 12, 15, 16
SAP 2:
One copy: 2, 3, 4, 7, 8, 11, 12, 15, 16
Skipping recombination in the following individuals: 5, 6, 9, 10, 13, 14
Simulation finished. Time used: 0.059 seconds
---------------
Results:
count.all fraction.all average.all longest.all count.rand
2.000 1.956 28.011 35.657 1.000
fraction.rand average.rand longest.rand length.dis rank.dis
0.711 20.365 20.365 35.657 1.000
zeroprob
0.000
Total time used: 0.067 seconds.
>
>
> #### Let us look at a different relationship: Half first cousins.
> # Two such cousins will on average share 1/8 = 12.5% of the autosome.
>
> z = halfCousinPed(1)
> plot(z)
> res = IBDsim(z, sims=1, query=list('1'=8:9))
---------------
Performing unconditional simulation
Mode: autosomal
Recombination model: chi square renewal model
Number of simulations: 1
Skipping recombination in the following individuals: 2, 3, 5, 7
Simulation finished. Time used: 0.016 seconds
---------------
Results:
count.all fraction.all average.all longest.all zeroprob
12.000 7.516 17.940 63.398 0.000
Total time used: 0.021 seconds.
> res$stats
[,1]
count.all 12.000000
fraction.all 7.515842
average.all 17.940307
longest.all 63.398452
>
> # visualizing the spread in total IBD sharing and the number of segments:
>
> IBD_percent = res$stats['fraction.all', ]
> IBD_count = res$stats['count.all', ]
> hist(IBD_percent)
> hist(IBD_count)
>
>
>
>
>
> dev.off()
null device
1
>
|