Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
KCsmart Comparative wrapperDescriptionWrapper function that calculates the sample point matrix collection from the aCGH data. The sample point matrix collection is used in the comparative version of KCsmart. UsagecalcSpmCollection(data, mirrorLocs, cl=NULL, data2=NULL, sigma=1000000, sampleDensity=50000, maxmem=1000, verbose=F, doChecks=T, old=F) Arguments
DetailsThe input can either consist of a single data set and a class vector or two separate datasets. In the latter case a class vector will be created assigning each data set to its own class. 'data' can be in cghRaw (CGHbase), DNAcopy or in data.frame format. When using the latter, the data.frame must have the following two columns: 'chrom' stating the chromosome the probe is located on, 'maploc' describing the position on the chromosome of the probe. The remainder of the data.frame will be interpreted as sample data points. The row names of that data will be used as probe names (when available). Important note: the data can not contain any missing values. If your data includes missing values you will need to preprocess (for example impute) it using other software solutions. The mirror locations for Homo Sapiens and Mus Musculus are provided in the package. These can be loaded using data(hsMirrorLocs) and data(mmMirrorLocs) respectively. The 'mirrorLocs' object is a list with vectors containing the start, centromere (optional) and end of each chromosome as the list elements. Additionally it should contain an attribute 'chromNames' listing the chromosome names of each respective list element. 'sigma' defines the kernel width of the kernel used to convolute the data. 'sampleDensity' defines the resolution of the sample point matrix to be calculated. A sampleDensity of 50000 would correspond to a sample point every 50k base pairs. 'old' can be used if you want to reproduce data that was generated with old (pre 2.9.0) versions of KCsmart, for any new analyses we recommend this flag to be set to false ValueReturns a sample point matrix collection object. The object has several slots of which the 'data' slot contains a matrix with the kernel smoothed estimates of all samples. The sample point matrix collection contains the following additional slots: cl: A class vector indicating which samples belong to which class. annotation: The annotation (containing the chromosome and position on the chromosome) for the sample points in the 'data' slot The other slots just represent the parameters used to calculate the sample point matrix collection. Use 'compareSpmCollection' to get a 'compKc' object for which the significant regions can be calculated using 'getSigRegionsCompKC'. Author(s)Jorma de Ronde See Also
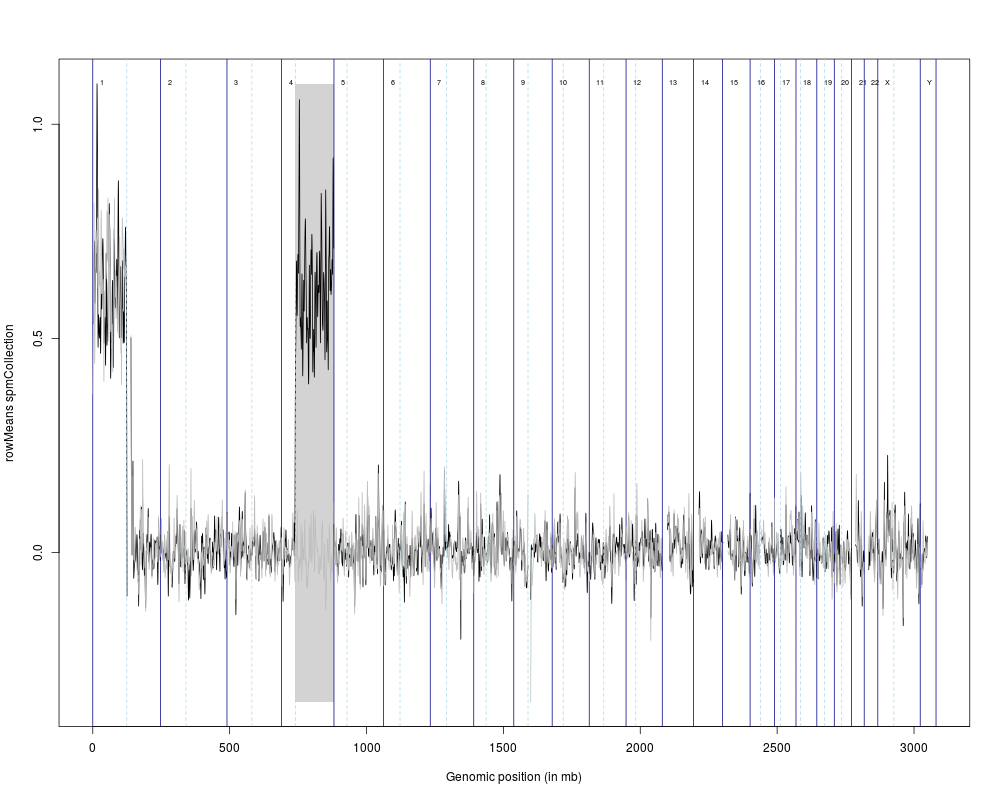
Examplesdata(hsSampleData) data(hsMirrorLocs) spmc1mb <- calcSpmCollection(hsSampleData, hsMirrorLocs, cl=c(rep(0,10),rep(1,10))) spmcc1mb <- compareSpmCollection(spmc1mb, nperms=3) spmcc1mbSigRegions <- getSigRegionsCompKC(spmcc1mb) plot(spmcc1mb, sigRegions=spmcc1mbSigRegions) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(KCsmart)
Loading required package: siggenes
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: multtest
Loading required package: splines
Loading required package: KernSmooth
KernSmooth 2.23 loaded
Copyright M. P. Wand 1997-2009
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/KCsmart/calcSpmCollection.Rd_%03d_medium.png", width=480, height=480)
> ### Name: calcSpmCollection
> ### Title: KCsmart Comparative wrapper
> ### Aliases: calcSpmCollection
> ### Keywords: manip
>
> ### ** Examples
>
> data(hsSampleData)
> data(hsMirrorLocs)
>
> spmc1mb <- calcSpmCollection(hsSampleData, hsMirrorLocs, cl=c(rep(0,10),rep(1,10)))
[1] "Mirror locations looking fine"
Processing sample 1 / 20 Processing sample 2 / 20 Processing sample 3 / 20 Processing sample 4 / 20 Processing sample 5 / 20 Processing sample 6 / 20 Processing sample 7 / 20 Processing sample 8 / 20 Processing sample 9 / 20 Processing sample 10 / 20 Processing sample 11 / 20 Processing sample 12 / 20 Processing sample 13 / 20 Processing sample 14 / 20 Processing sample 15 / 20 Processing sample 16 / 20 Processing sample 17 / 20 Processing sample 18 / 20 Processing sample 19 / 20 Processing sample 20 / 20
> spmcc1mb <- compareSpmCollection(spmc1mb, nperms=3)
Warning messages:
1: There are 3294 genes with at least one missing expression value.
The NAs are replaced by the gene-wise mean.
2: 3294 of the 3294 genes with at least one NA have no and 0 have one non-missing expression value.
All these 3294 genes are removed, and their d-values are set to NA.
> spmcc1mbSigRegions <- getSigRegionsCompKC(spmcc1mb)
>
> plot(spmcc1mb, sigRegions=spmcc1mbSigRegions)
>
>
>
>
>
>
> dev.off()
null device
1
>
|