Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Query the subgraph of a given KEGG graph with Entrez GeneID (s)DescriptionGiven a list of genes (identified by Entrez GeneID), the function subsets the given KEGG gragh of the genes as nodes (and maintaining all the edges between). UsagequeryKEGGsubgraph(geneids, graph, organism = "hsa", addmissing = FALSE) Arguments
DetailsThis function solves the questions like 'How is the list of gene interact with each other in the context of pathways?' Limited by the If 'addmissing' is set to ValueA subgraph with nodes representing genes and edges representing interactions. Author(s)Jitao David Zhang <jitao_david.zhang@roche.com> See Also
Examples
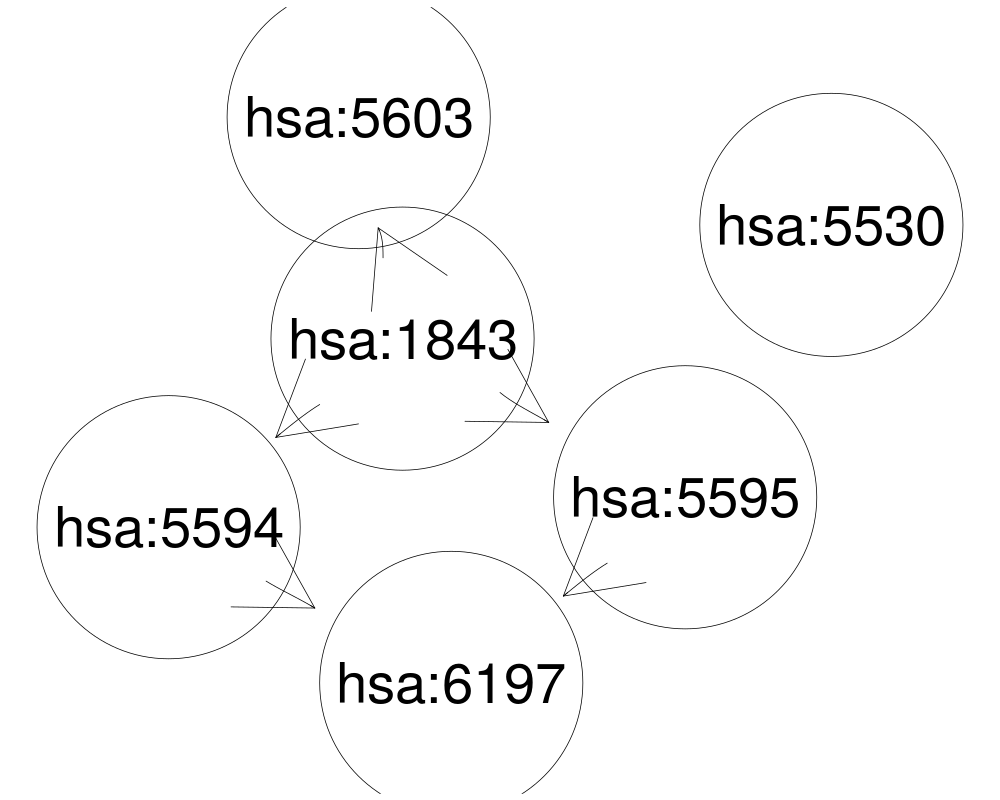
sfile <- system.file("extdata/hsa04010.xml",package="KEGGgraph")
gR <- parseKGML2Graph(sfile,expandGenes=TRUE)
geneids <- c(5594, 5595, 6197, 5603, 1843,5530, 5603)
sub <- queryKEGGsubgraph(geneids, gR)
if(require(Rgraphviz) && interactive()) {
plot(sub, "neato")
}
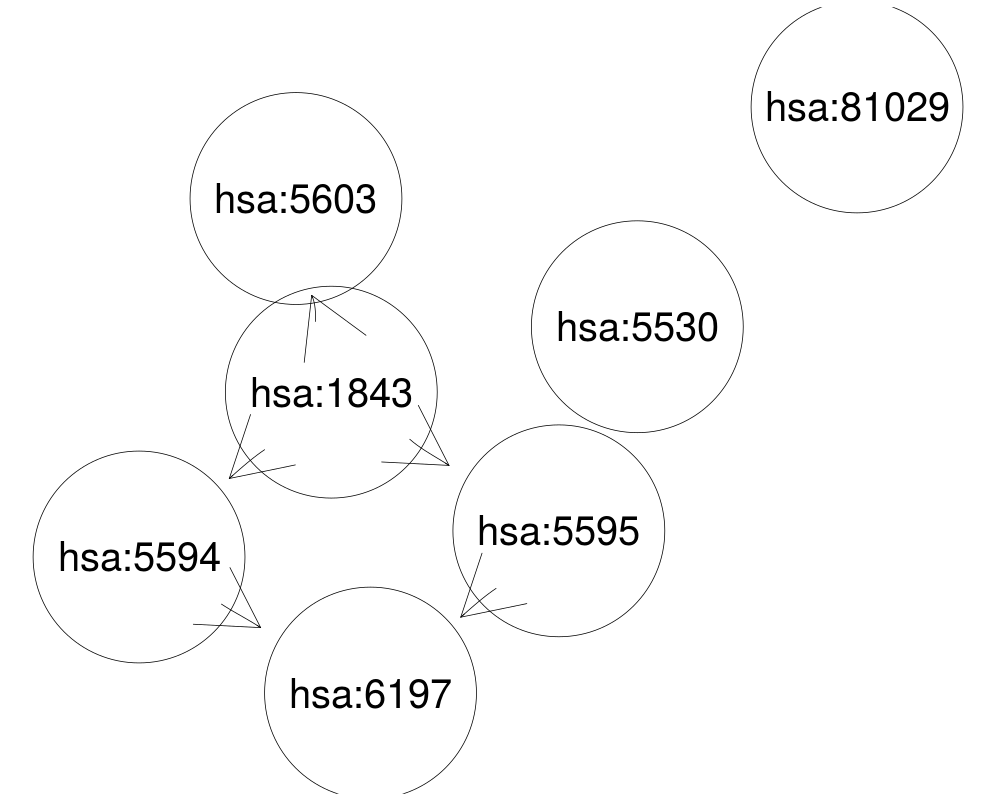
## add missing nodes
list2 <- c(geneids, 81029)
sub2 <- queryKEGGsubgraph(list2, gR,addmissing=TRUE)
if(require(Rgraphviz) && interactive()) {
plot(sub2, "neato")
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(KEGGgraph)
Attaching package: 'KEGGgraph'
The following object is masked from 'package:graphics':
plot
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/KEGGgraph/queryKEGGsubgraph.Rd_%03d_medium.png", width=480, height=480)
> ### Name: queryKEGGsubgraph
> ### Title: Query the subgraph of a given KEGG graph with Entrez GeneID (s)
> ### Aliases: queryKEGGsubgraph
>
> ### ** Examples
>
> sfile <- system.file("extdata/hsa04010.xml",package="KEGGgraph")
> gR <- parseKGML2Graph(sfile,expandGenes=TRUE)
> geneids <- c(5594, 5595, 6197, 5603, 1843,5530, 5603)
> sub <- queryKEGGsubgraph(geneids, gR)
> #if(require(Rgraphviz) && interactive()) {
> plot(sub, "neato")
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
> #}
>
> ## add missing nodes
> list2 <- c(geneids, 81029)
> sub2 <- queryKEGGsubgraph(list2, gR,addmissing=TRUE)
> #if(require(Rgraphviz) && interactive()) {
> plot(sub2, "neato")
> #}
>
>
>
>
>
> dev.off()
null device
1
>
|