Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Estimates individual ancestry coefficients and ancestral allele frequencies.Description
Usage
snmf (input.file, K,
project = "continue",
repetitions = 1, CPU = 1,
alpha = 10, tolerance = 0.00001, entropy = FALSE, percentage = 0.05,
I, iterations = 200, ploidy = 2, seed = -1, Q.input.file)
Arguments
Value
The following methods can be applied to the object of class snmfProject:
Author(s)Eric Frichot ReferencesFrichot E, Mathieu F, Trouillon T, Bouchard G, Francois O. (2014). Fast and Efficient Estimation of Individual Ancestry Coefficients. Genetics, 194(4): 973–983. See Also
Examples
### Example of analyses using snmf ###
# creation of the genotype file, genotypes.geno.
# It contains 400 SNPs for 50 individuals.
data("tutorial")
write.geno(tutorial.R, "genotypes.geno")
################
# runs of snmf #
################
# main options, K: (the number of ancestral populations),
# entropy: calculate the cross-entropy criterion,
# CPU: the number of CPUs.
# Runs with K between 1 and 5 with cross-entropy and 2 repetitions.
project = NULL
project = snmf("genotypes.geno", K=1:10, entropy = TRUE, repetitions = 10,
project = "new")
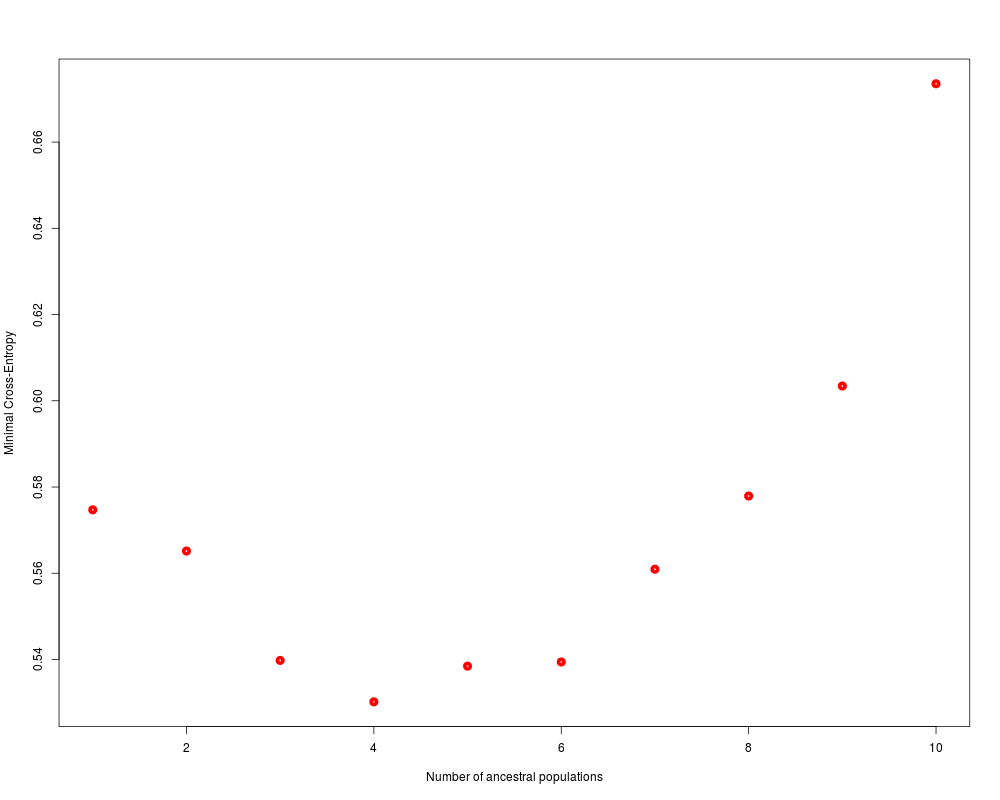
# plot cross-entropy criterion of all runs of the project
plot(project, lwd = 5, col = "red", pch=1)
# get the cross-entropy of each run for K = 4
ce = cross.entropy(project, K = 4)
# select the run with the lowest cross-entropy
best = which.min(ce)
# plot the best run for K = 4 (ancestry coefficients).
barplot(t(Q(project, K = 4, run = best)))
###################
# Post-treatments #
###################
# show the project
show(project)
# summary of the project
summary(project)
# get the cross-entropy for all runs for K = 4
ce = cross.entropy(project, K = 4)
# get the cross-entropy for the 2nd run for K = 4
ce = cross.entropy(project, K = 4, run = 2)
# get the ancestral genotype frequency matrix, G, for the 2nd run for K = 4.
res = G(project, K = 4, run = 2)
#############################
# Advanced snmf run options #
#############################
# Q.input.file: init a run with a given ancestry coefficient matrix Q.
# Here, it is initialized with the Q matrix from the first run with K=4
project = snmf("genotypes.geno", K = 4,
Q.input.file = "./genotypes.snmf/K4/run1/genotypes_r1.4.Q")
# I: init the Q matrix of a run from a smaller run with 100 randomly chosen
# SNPs.
project = snmf("genotypes.geno", K = 4, I = 100)
# CPU: run snmf with 2 CPUs.
project = snmf("genotypes.geno", K = 4, CPU=2)
# percentage: run snmf and calculate the cross-entropy criterion with 10% of
# masked genotypes, instead of 5% of masked genotypes.
project = snmf("genotypes.geno", K = 4, entropy= TRUE, percentage = 0.1)
# seed: choose the seed to init the randomization.
project = snmf("genotypes.geno", K = 4, seed=42)
# alpha: choose the regularization parameter.
project = snmf("genotypes.geno", K = 4, alpha = 100)
# tolerance: choose the tolerance parameter.
project = snmf("genotypes.geno", K = 4, tolerance = 0.0001)
##########################
# Manage an snmf project #
##########################
# All the runs of snmf for a given file are
# automatically saved into a snmf project directory and a file.
# The name of the snmfProject file is the same name as
# the name of the input file with a .snmfProject extension
# ("genotypes.snmfProject").
# The name of the snmfProject directory is the same name as
# the name of the input file with a .snmf extension ("genotypes.snmf/")
# There is only one snmf Project for each input file including all the runs.
# An snmfProject can be load in a different session.
project = load.snmfProject("genotypes.snmfProject")
# An snmfProject can be exported to be imported in another directory
# or in another computer
export.snmfProject("genotypes.snmfProject")
dir.create("test", showWarnings = TRUE)
#import
newProject = import.snmfProject("genotypes_snmfProject.zip", "test")
# combine projects
combinedProject = combine.snmfProject("genotypes.snmfProject", "test/genotypes.snmfProject")
# remove
remove.snmfProject("test/genotypes.snmfProject")
# An snmfProject can be erased.
# Caution: All the files associated with the project will be removed.
remove.snmfProject("genotypes.snmfProject")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(LEA)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/LEA/main_sNMF.Rd_%03d_medium.png", width=480, height=480)
> ### Name: snmf
> ### Title: Estimates individual ancestry coefficients and ancestral allele
> ### frequencies.
> ### Aliases: snmf plot,snmfProject-method cross.entropy,snmfProject-method
> ### load.snmfProject load.snmfProject,character-method remove.snmfProject
> ### remove.snmfProject,character-method import.snmfProject
> ### import.snmfProject,character-method export.snmfProject
> ### export.snmfProject,character-method combine.snmfProject
> ### combine.snmfProject,character,character-method
> ### show,snmfProject-method show,snmfClass-method
> ### summary,snmfProject-method Q,snmfProject-method G,snmfProject-method
> ### Keywords: snmf tutorial
>
> ### ** Examples
>
> ### Example of analyses using snmf ###
>
> # creation of the genotype file, genotypes.geno.
> # It contains 400 SNPs for 50 individuals.
> data("tutorial")
> write.geno(tutorial.R, "genotypes.geno")
[1] "genotypes.geno"
>
> ################
> # runs of snmf #
> ################
>
> # main options, K: (the number of ancestral populations),
> # entropy: calculate the cross-entropy criterion,
> # CPU: the number of CPUs.
>
> # Runs with K between 1 and 5 with cross-entropy and 2 repetitions.
> project = NULL
> project = snmf("genotypes.geno", K=1:10, entropy = TRUE, repetitions = 10,
+ project = "new")
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] 541156239
[1] "*************************************"
[1] "* create.dataset *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-s (seed random init) 541156239
-r (percentage of masked data) 0.05
-x (genotype file in .geno format) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-o (output file in .geno format) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
Write genotype file with masked data, /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
[1] "*************************************"
[1] "* sNMF K = 1 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 1
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
Least-square error: 6669.480084
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 1
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run1/genotypes_r1.1.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.576028
Cross-Entropy (masked data): 0.687661
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 2 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 2
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 9896145806223
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[===================]
Number of iterations: 50
Least-square error: 6114.399730
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 2
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K2/run1/genotypes_r1.2.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.517816
Cross-Entropy (masked data): 0.661273
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 3 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 3
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[===============================]
Number of iterations: 82
Least-square error: 5701.311254
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 3
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K3/run1/genotypes_r1.3.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.476134
Cross-Entropy (masked data): 0.641755
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 4 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 4
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[==============================]
Number of iterations: 79
Least-square error: 5418.876217
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 4
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K4/run1/genotypes_r1.4.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.450315
Cross-Entropy (masked data): 0.650196
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 5 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 5
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[=========================]
Number of iterations: 68
Least-square error: 5225.566484
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 5
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K5/run1/genotypes_r1.5.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.430297
Cross-Entropy (masked data): 0.698477
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 6 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 6
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 4611686018968544143
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[================================]
Number of iterations: 86
Least-square error: 5058.327447
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 6
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K6/run1/genotypes_r1.6.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.415832
Cross-Entropy (masked data): 0.709669
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 7 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 7
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[================]
Number of iterations: 43
Least-square error: 4934.204204
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 7
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K7/run1/genotypes_r1.7.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.4044
Cross-Entropy (masked data): 0.708292
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 8 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 8
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[=======================================]
Number of iterations: 104
Least-square error: 4745.499255
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 8
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K8/run1/genotypes_r1.8.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.386626
Cross-Entropy (masked data): 0.765435
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 9 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 9
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[===========================]
Number of iterations: 73
Least-square error: 4638.974246
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 9
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K9/run1/genotypes_r1.9.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.375517
Cross-Entropy (masked data): 0.772626
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] "*************************************"
[1] "* sNMF K = 10 repetition 1 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 10
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 541156239
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
Main algorithm:
[ ]
[===========]
Number of iterations: 29
Least-square error: 4490.121237
Write individual ancestry coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.Q: OK.
Write ancestral allele frequency coefficient file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.G: OK.
[1] "*************************************"
[1] "* cross-entropy estimation *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 10
-x (genotype file) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-q (individual admixture) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.Q
-g (ancestral frequencies) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K10/run1/genotypes_r1.10.G
-i (with masked genotypes) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
- diploid
Cross-Entropy (all data): 0.36707
Cross-Entropy (masked data): 0.838762
The project is saved into :
genotypes.snmfProject
To load the project, use:
project = load.snmfProject("genotypes.snmfProject")
To remove the project, use:
remove.snmfProject("genotypes.snmfProject")
[1] 1587990360
[1] "*************************************"
[1] "* create.dataset *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-s (seed random init) 1587990360
-r (percentage of masked data) 0.05
-x (genotype file in .geno format) /home/ddbj/DataUpdator-rgm3/target/genotypes.geno
-o (output file in .geno format) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
Write genotype file with masked data, /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno: OK.
[1] "*************************************"
[1] "* sNMF K = 1 repetition 2 *"
[1] "*************************************"
summary of the options:
-n (number of individuals) 50
-L (number of loci) 400
-K (number of ancestral pops) 1
-x (input file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno
-q (individual admixture file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run2/genotypes_r2.1.Q
-g (ancestral frequencies file) /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/K1/run2/genotypes_r2.1.G
-i (number max of iterations) 200
-a (regularization parameter) 10
-s (seed random init) 1587990360
-e (tolerance error) 1E-05
-p (number of processes) 1
- diploid
Read genotype file /home/ddbj/DataUpdator-rgm3/target/genotypes.snmf/masked/genotypes_I.geno:
|