Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Clone tracking plotDescriptionCreates line plot tracking amino acid frequencies across multiple samples UsagecloneTrack(sequence.matrix, map = "none", productive.aa, label = "none", track = "none", unassigned = TRUE) Arguments
DetailsThe plot is made using the package ggplot2 and can be reformatted using ggplot2 functions. See examples below. ValueReturns a line plot showing the amino acid frequencies across multiple samples in the sequence matrix where each line represents one unique sequence. See AlsoAn excellent resource for examples on how to reformat a ggplot can be found in the R Graphics Cookbook online (http://www.cookbook-r.com/Graphs/). Examples
file.path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeq")
file.list <- readImmunoSeq(path = file.path)
productive.aa <- productiveSeq(file.list = file.list, aggregate = "aminoAcid")
top.freq <- topFreq(productive.aa = productive.aa, percent = 0.1)
sequence.matrix <- seqMatrix(productive.aa = productive.aa, sequences = top.freq$aminoAcid)
# Track clones without mapping or tracking specific sequences
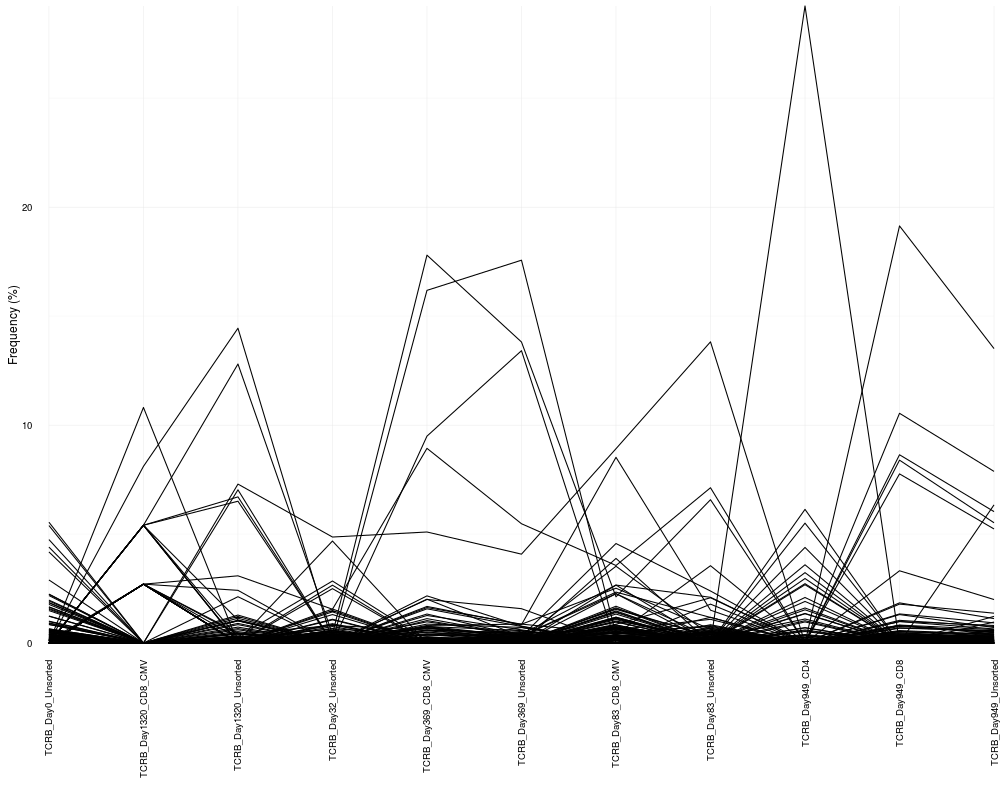
cloneTrack(sequence.matrix = sequence.matrix)
# Track top 20 clones mapping to the CD4 and CD8 samples
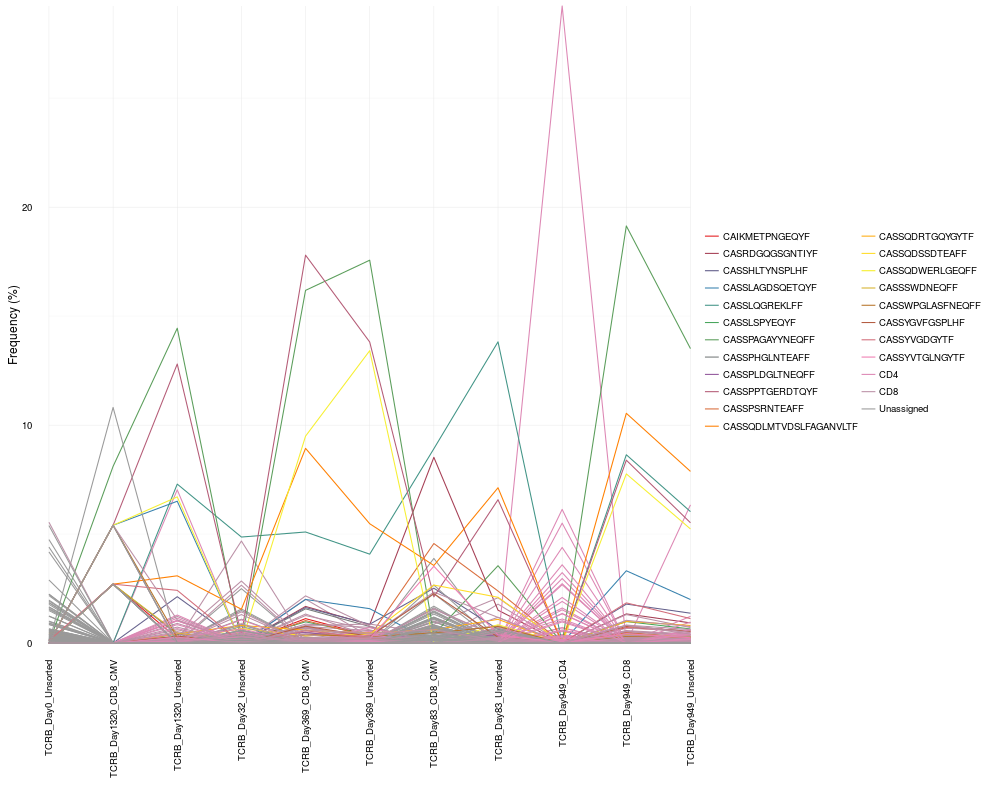
cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
track = top.freq$aminoAcid[1:20], unassigned = TRUE)
# Track the top 10 clones from top.freq
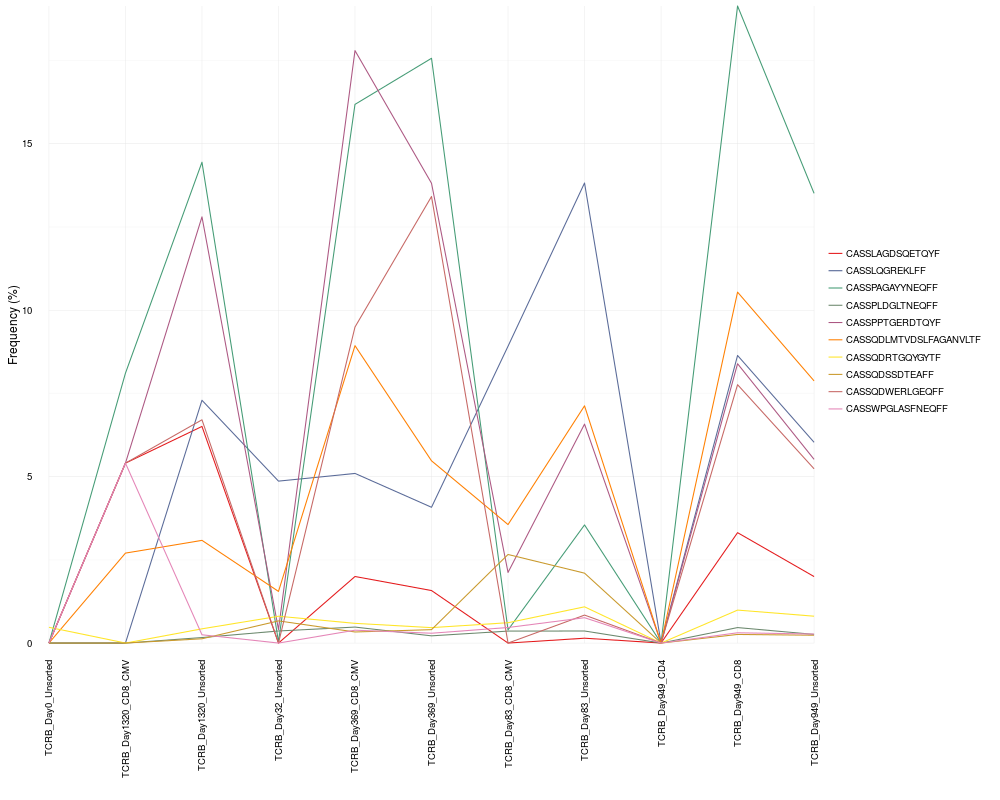
cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
track = top.freq$aminoAcid[1:10], unassigned = FALSE)
# Track clones mapping to the CD4 and CD8 samples while ignoring all others
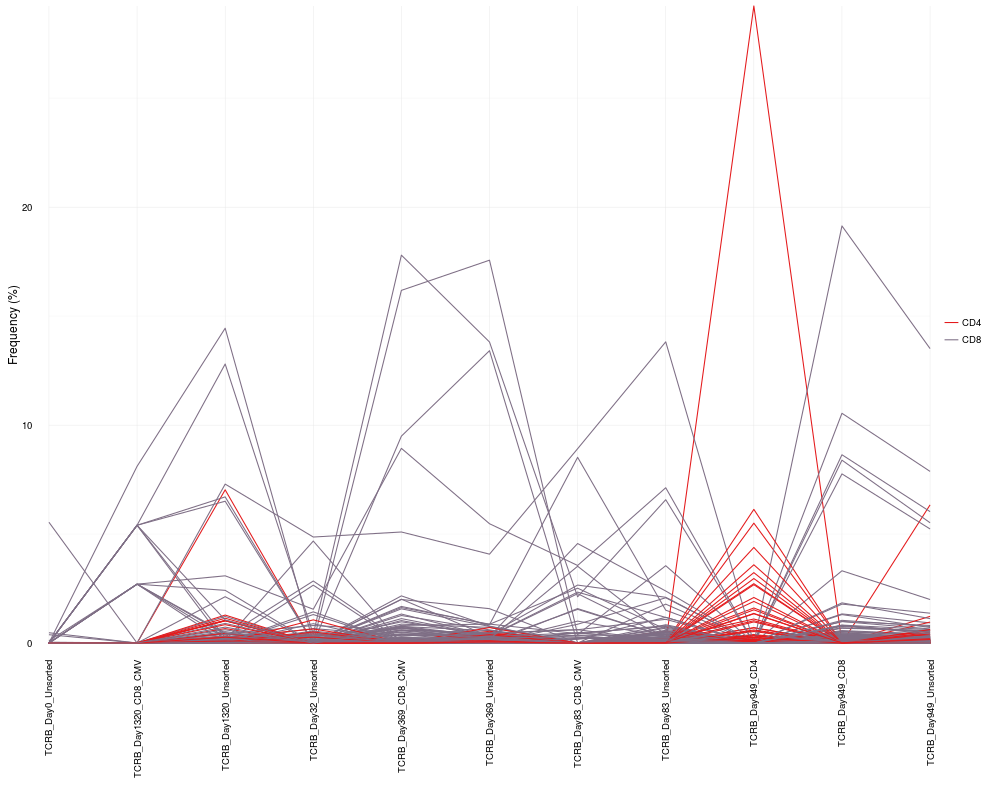
cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
unassigned = FALSE)
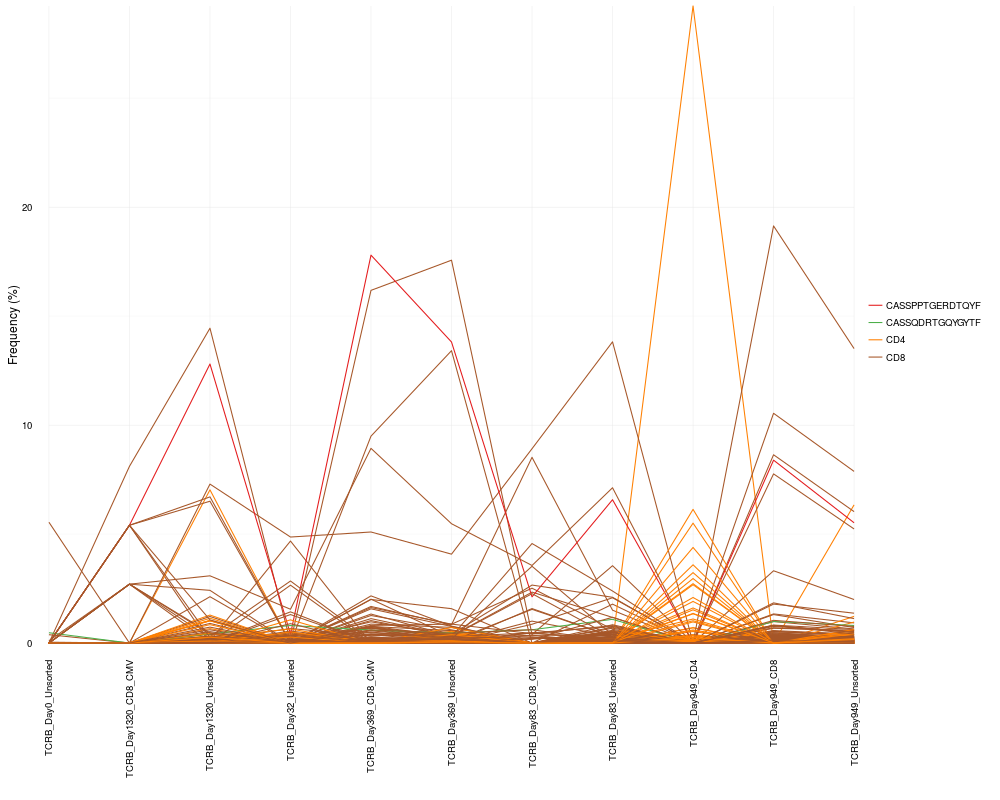
# Track clones mapping to the CD4 and CD8 samples and track 2 specific sequences
cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
track = c("CASSPPTGERDTQYF", "CASSQDRTGQYGYTF"), unassigned = FALSE)
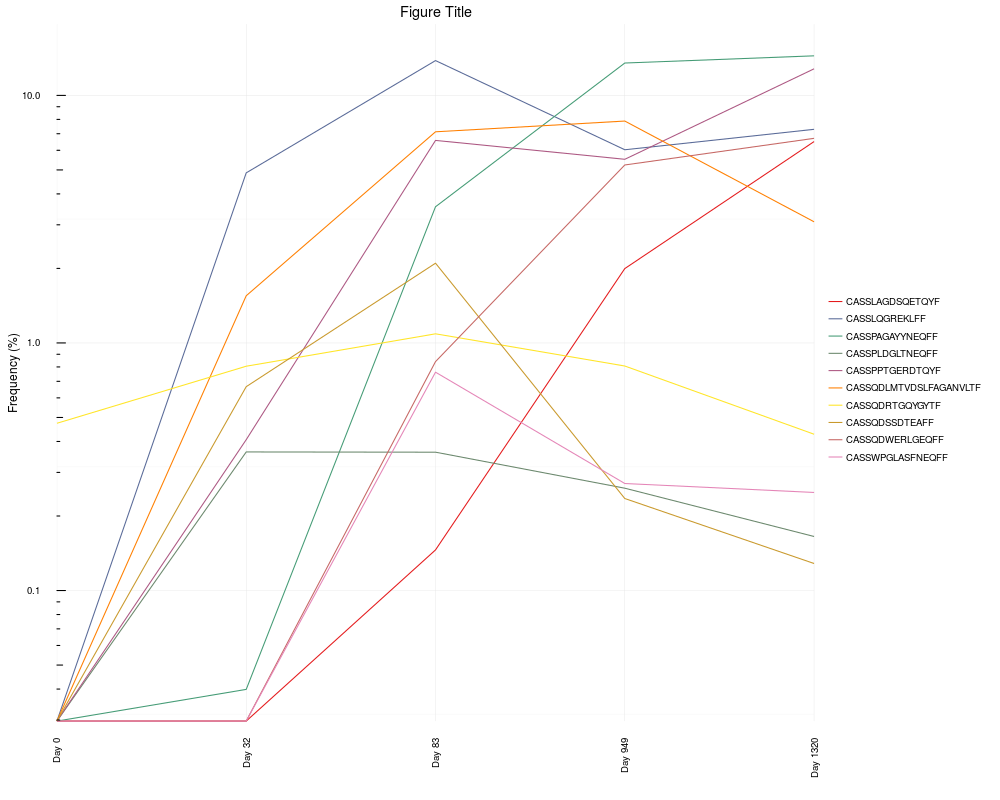
# Reorder the x axis, change the axis labels, convert to log scale, and add title
x.limits <- c("TCRB_Day0_Unsorted", "TCRB_Day32_Unsorted",
"TCRB_Day83_Unsorted", "TCRB_Day949_Unsorted", "TCRB_Day1320_Unsorted")
sequence.matrix <- sequence.matrix[ ,c("aminoAcid", x.limits)]
clone.track <- cloneTrack(sequence.matrix = sequence.matrix,
productive.aa = productive.aa, track = top.freq$aminoAcid[1:10], unassigned = FALSE)
x.labels <- c("Day 0", "Day 32", "Day 83", "Day 949", "Day 1320")
clone.track +
ggplot2::scale_x_discrete(expand = c(0,0), labels = x.labels) +
ggplot2::scale_y_log10() + ggplot2::annotation_logticks(sides = "l") +
ggplot2::ggtitle("Figure Title")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(LymphoSeq)
Loading required package: LymphoSeqDB
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/LymphoSeq/cloneTrack.Rd_%03d_medium.png", width=480, height=480)
> ### Name: cloneTrack
> ### Title: Clone tracking plot
> ### Aliases: cloneTrack
>
> ### ** Examples
>
> file.path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeq")
>
> file.list <- readImmunoSeq(path = file.path)
| | | 0% | |====== | 9% | |============= | 18% | |=================== | 27% | |========================= | 36% | |================================ | 45% | |====================================== | 55% | |============================================= | 64% | |=================================================== | 73% | |========================================================= | 82% | |================================================================ | 91% | |======================================================================| 100%
>
> productive.aa <- productiveSeq(file.list = file.list, aggregate = "aminoAcid")
| | | 0% | |====== | 9% | |============= | 18% | |=================== | 27% | |========================= | 36% | |================================ | 45% | |====================================== | 55% | |============================================= | 64% | |=================================================== | 73% | |========================================================= | 82% | |================================================================ | 91% | |======================================================================| 100%
>
> top.freq <- topFreq(productive.aa = productive.aa, percent = 0.1)
>
> sequence.matrix <- seqMatrix(productive.aa = productive.aa, sequences = top.freq$aminoAcid)
>
> # Track clones without mapping or tracking specific sequences
> cloneTrack(sequence.matrix = sequence.matrix)
>
> # Track top 20 clones mapping to the CD4 and CD8 samples
> cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
+ map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
+ track = top.freq$aminoAcid[1:20], unassigned = TRUE)
>
> # Track the top 10 clones from top.freq
> cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
+ track = top.freq$aminoAcid[1:10], unassigned = FALSE)
>
> # Track clones mapping to the CD4 and CD8 samples while ignoring all others
> cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
+ map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
+ unassigned = FALSE)
>
> # Track clones mapping to the CD4 and CD8 samples and track 2 specific sequences
> cloneTrack(sequence.matrix = sequence.matrix, productive.aa = productive.aa,
+ map = c("TCRB_Day949_CD4", "TCRB_Day949_CD8"), label = c("CD4", "CD8"),
+ track = c("CASSPPTGERDTQYF", "CASSQDRTGQYGYTF"), unassigned = FALSE)
>
> # Reorder the x axis, change the axis labels, convert to log scale, and add title
> x.limits <- c("TCRB_Day0_Unsorted", "TCRB_Day32_Unsorted",
+ "TCRB_Day83_Unsorted", "TCRB_Day949_Unsorted", "TCRB_Day1320_Unsorted")
>
> sequence.matrix <- sequence.matrix[ ,c("aminoAcid", x.limits)]
>
> clone.track <- cloneTrack(sequence.matrix = sequence.matrix,
+ productive.aa = productive.aa, track = top.freq$aminoAcid[1:10], unassigned = FALSE)
>
> x.labels <- c("Day 0", "Day 32", "Day 83", "Day 949", "Day 1320")
>
> clone.track +
+ ggplot2::scale_x_discrete(expand = c(0,0), labels = x.labels) +
+ ggplot2::scale_y_log10() + ggplot2::annotation_logticks(sides = "l") +
+ ggplot2::ggtitle("Figure Title")
Scale for 'x' is already present. Adding another scale for 'x', which will
replace the existing scale.
Scale for 'y' is already present. Adding another scale for 'y', which will
replace the existing scale.
>
>
>
>
>
> dev.off()
null device
1
>
|