Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
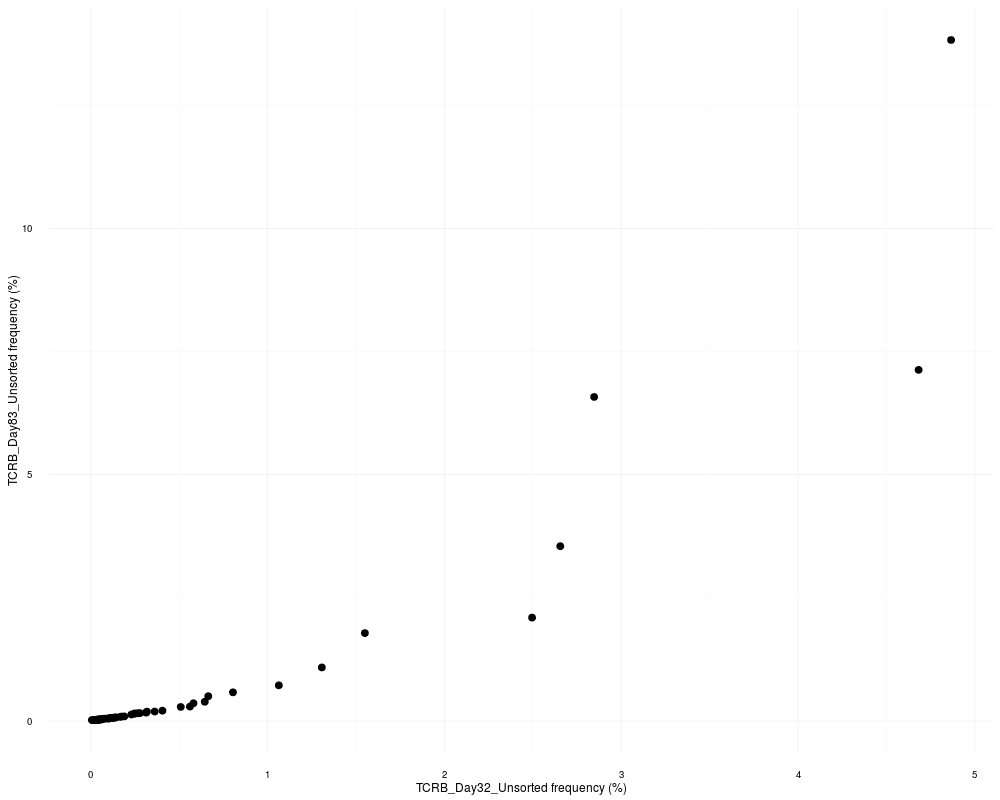
Common sequences plotDescriptionCreates a scatter plot of just the sequences in common between two samples. UsagecommonSeqsPlot(sample1, sample2, productive.aa) Arguments
DetailsThe plot is made using the package ggplot2 and can be reformatted using ggplot2 functions. See examples below. ValueReturns a frequency scatter plot of two samples showing only the shared sequences. See AlsoAn excellent resource for examples on how to reformat a ggplot can be found in the R Graphics Cookbook online (http://www.cookbook-r.com/Graphs/). Examples
file.path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeq")
file.list <- readImmunoSeq(path = file.path)
productive.aa <- productiveSeq(file.list = file.list, aggregate = "aminoAcid")
commonSeqsPlot("TCRB_Day32_Unsorted", "TCRB_Day83_Unsorted",
productive.aa = productive.aa)
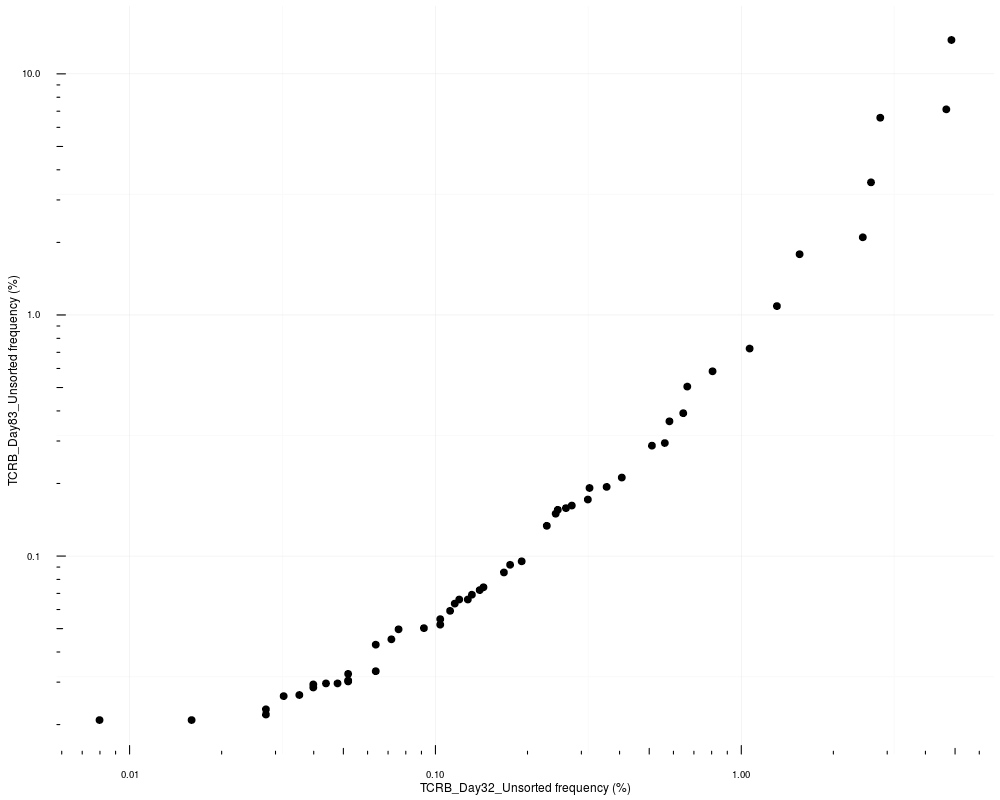
# Change the X and Y axises to log-10 scale
commonSeqsPlot("TCRB_Day32_Unsorted", "TCRB_Day83_Unsorted",
productive.aa = productive.aa) +
ggplot2::scale_x_log10() +
ggplot2::scale_y_log10() +
ggplot2::annotation_logticks(sides = "bl")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(LymphoSeq)
Loading required package: LymphoSeqDB
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/LymphoSeq/commonSeqsPlot.Rd_%03d_medium.png", width=480, height=480)
> ### Name: commonSeqsPlot
> ### Title: Common sequences plot
> ### Aliases: commonSeqsPlot
>
> ### ** Examples
>
> file.path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeq")
>
> file.list <- readImmunoSeq(path = file.path)
| | | 0% | |====== | 9% | |============= | 18% | |=================== | 27% | |========================= | 36% | |================================ | 45% | |====================================== | 55% | |============================================= | 64% | |=================================================== | 73% | |========================================================= | 82% | |================================================================ | 91% | |======================================================================| 100%
>
> productive.aa <- productiveSeq(file.list = file.list, aggregate = "aminoAcid")
| | | 0% | |====== | 9% | |============= | 18% | |=================== | 27% | |========================= | 36% | |================================ | 45% | |====================================== | 55% | |============================================= | 64% | |=================================================== | 73% | |========================================================= | 82% | |================================================================ | 91% | |======================================================================| 100%
>
> commonSeqsPlot("TCRB_Day32_Unsorted", "TCRB_Day83_Unsorted",
+ productive.aa = productive.aa)
>
> # Change the X and Y axises to log-10 scale
> commonSeqsPlot("TCRB_Day32_Unsorted", "TCRB_Day83_Unsorted",
+ productive.aa = productive.aa) +
+ ggplot2::scale_x_log10() +
+ ggplot2::scale_y_log10() +
+ ggplot2::annotation_logticks(sides = "bl")
>
>
>
>
>
> dev.off()
null device
1
>
|