Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Finds monoisotopic peaks in a MassPeaks object.DescriptionThis method looks for monoisotopic peaks in peak list data
(represented by a Usage## S4 method for signature 'MassPeaks' monoisotopicPeaks(object, minCor=0.95, tolerance=1e-4, distance=1.00235, size=3L:10L) ## S4 method for signature 'list' monoisotopicPeaks(object, ...) Arguments
ValueReturns a Author(s)Sebastian Gibb mail@sebastiangibb.de ReferencesK. Park, J.Y. Yoon, S. Lee, E. Paek, H. Park, H.J. Jung, and S.W. Lee. 2008. Isotopic peak intensity ratio based algorithm for determination of isotopic clusters and monoisotopic masses of polypeptides from high-resolution mass spectrometric data. Analytical Chemistry, 80: 7294-7303. E.J. Breen, F.G. Hopwood, K.L. Williams, and M.R. Wilkins. 2000. Automatic poisson peak harvesting for high throughput protein identification. Electrophoresis 21: 2243-2251. See Also
Website: http://strimmerlab.org/software/maldiquant/ Examples
## load package
library("MALDIquant")
## create example peaks
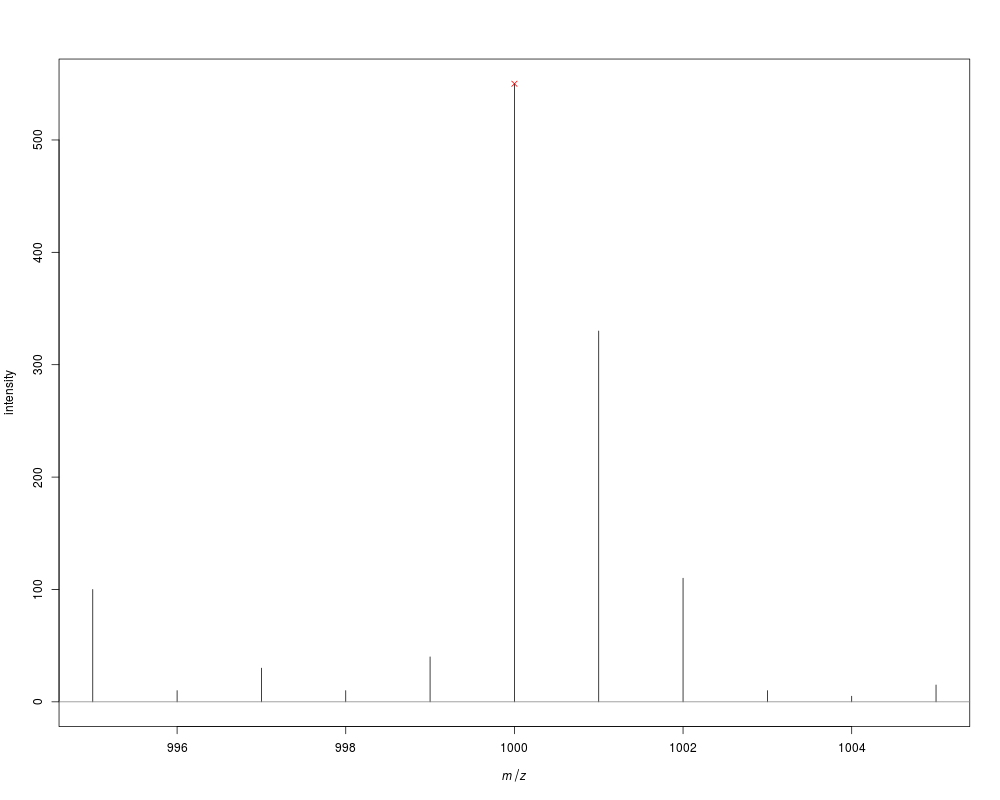
p <- createMassPeaks(mass=995:1005,
intensity=c(100, 10, 30, 10, 40, # noise
550, 330, 110, 10, # isotopic pattern
5, 15)) # more noise
m <- monoisotopicPeaks(p)
as.matrix(m)
## plot the peaks and mark the monoisotopic one
plot(p)
points(m, col=2, pch=4)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MALDIquant)
This is MALDIquant version 1.15
Quantitative Analysis of Mass Spectrometry Data
See '?MALDIquant' for more information about this package.
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/MALDIquant/monoisotopicPeaks-methods.Rd_%03d_medium.png", width=480, height=480)
> ### Name: monoisotopicPeaks-methods
> ### Title: Finds monoisotopic peaks in a MassPeaks object.
> ### Aliases: monoisotopicPeaks monoisotopicPeaks,MassPeaks-method
> ### monoisotopicPeaks,list-method
> ### Keywords: methods
>
> ### ** Examples
>
> ## load package
> library("MALDIquant")
>
> ## create example peaks
> p <- createMassPeaks(mass=995:1005,
+ intensity=c(100, 10, 30, 10, 40, # noise
+ 550, 330, 110, 10, # isotopic pattern
+ 5, 15)) # more noise
> m <- monoisotopicPeaks(p)
> as.matrix(m)
mass intensity
[1,] 1000 550
>
> ## plot the peaks and mark the monoisotopic one
> plot(p)
> points(m, col=2, pch=4)
>
>
>
>
>
> dev.off()
null device
1
>
|