Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Plotting fixed effects for all genes for a single combination of factorsDescriptionCalculates and plots posterior means with 95% credible intervals for specified fixed effects (or their combination) for all genes UsageHPDplot(model, factors, factors2 = NULL, ylimits = NULL, hpdtype = "w", inverse = F, jitter = 0, plot = T, grid = T, zero = T, ...) Arguments
DetailsUse summary(MCMCglmm object) first to see what fixed effect names are actually used in the output. For example, if summary shows: gene1:conditionheat , it is possible to specify factors="conditionheat" to plot only the effects of the heat. If a vector of several fixed effect names is given, for example: factors=c("timepointtwo","treatmentheat:timepointtwo") the function will plot the posterior mean and credible interval for the sum of these effects. If a second vector is also given, for example, ValueA plot or a table (plot = F). Use the function HPDpoints() if you need to add graphs to already existing plot. Author(s)Mikhail V. Matz, UT Austin <matz@utexas.edu> ReferencesMatz MV, Wright RM, Scott JG (2013) No Control Genes Required: Bayesian Analysis of qRT-PCR Data. PLoS ONE 8(8): e71448. doi:10.1371/journal.pone.0071448 Examples
# loading Cq data and amplification efficiencies
data(coral.stress)
data(amp.eff)
# extracting a subset of data
cs.short=subset(coral.stress, timepoint=="one")
genecolumns=c(5,6,16,17) # specifying columns corresponding to genes of interest
conditions=c(1:4) # specifying columns containing factors
# calculating molecule counts and reformatting:
dd=cq2counts(data=cs.short,genecols=genecolumns,
condcols=conditions,effic=amp.eff,Cq1=37)
# fitting the model
mm=mcmc.qpcr(
fixed="condition",
data=dd,
controls=c("nd5","rpl11"),
nitt=3000,burnin=2000 # remove this line when analyzing real data!
)
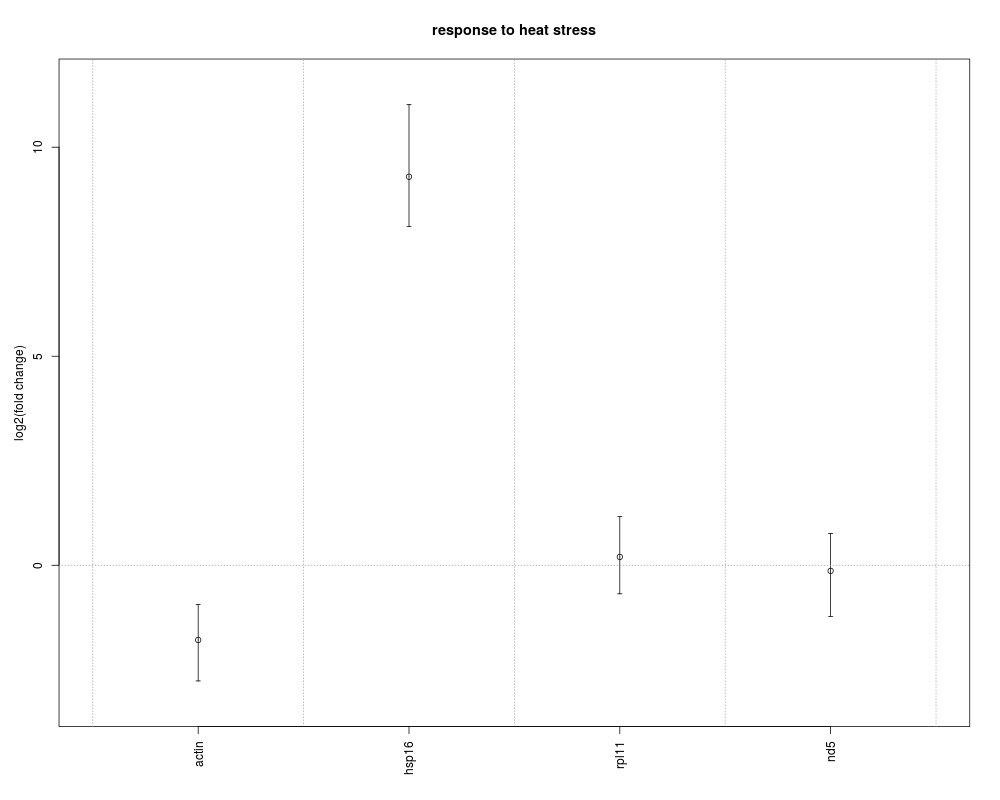
# plotting log2(fold change) in response to heat stress for all genes
HPDplot(model=mm,factors="conditionheat",main="response to heat stress")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MCMC.qpcr)
Loading required package: MCMCglmm
Loading required package: Matrix
Loading required package: coda
Loading required package: ape
Loading required package: ggplot2
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/MCMC.qpcr/HPDplot.Rd_%03d_medium.png", width=480, height=480)
> ### Name: HPDplot
> ### Title: Plotting fixed effects for all genes for a single combination of
> ### factors
> ### Aliases: HPDplot
>
> ### ** Examples
>
>
> # loading Cq data and amplification efficiencies
> data(coral.stress)
> data(amp.eff)
> # extracting a subset of data
> cs.short=subset(coral.stress, timepoint=="one")
>
> genecolumns=c(5,6,16,17) # specifying columns corresponding to genes of interest
> conditions=c(1:4) # specifying columns containing factors
>
> # calculating molecule counts and reformatting:
> dd=cq2counts(data=cs.short,genecols=genecolumns,
+ condcols=conditions,effic=amp.eff,Cq1=37)
>
> # fitting the model
> mm=mcmc.qpcr(
+ fixed="condition",
+ data=dd,
+ controls=c("nd5","rpl11"),
+ nitt=3000,burnin=2000 # remove this line when analyzing real data!
+ )
$PRIOR
$PRIOR$B
$PRIOR$B$mu
[1] 0 0 0 0 0 0 0 0
$PRIOR$B$V
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
[1,] 1e+08 0e+00 0e+00 0e+00 0e+00 0e+00 0.0000000 0.0000000
[2,] 0e+00 1e+08 0e+00 0e+00 0e+00 0e+00 0.0000000 0.0000000
[3,] 0e+00 0e+00 1e+08 0e+00 0e+00 0e+00 0.0000000 0.0000000
[4,] 0e+00 0e+00 0e+00 1e+08 0e+00 0e+00 0.0000000 0.0000000
[5,] 0e+00 0e+00 0e+00 0e+00 1e+08 0e+00 0.0000000 0.0000000
[6,] 0e+00 0e+00 0e+00 0e+00 0e+00 1e+08 0.0000000 0.0000000
[7,] 0e+00 0e+00 0e+00 0e+00 0e+00 0e+00 0.3663091 0.0000000
[8,] 0e+00 0e+00 0e+00 0e+00 0e+00 0e+00 0.0000000 0.3663091
$PRIOR$R
$PRIOR$R$V
[,1] [,2] [,3] [,4]
[1,] 1 0 0 0
[2,] 0 1 0 0
[3,] 0 0 1 0
[4,] 0 0 0 1
$PRIOR$R$nu
[1] 3.002
$PRIOR$G
$PRIOR$G$G1
$PRIOR$G$G1$V
[1] 1
$PRIOR$G$G1$nu
[1] 0
$FIXED
[1] "count~0+gene++gene:condition"
$RANDOM
[1] "~sample"
MCMC iteration = 0
Acceptance ratio for liability set 1 = 0.000438
Acceptance ratio for liability set 2 = 0.000419
Acceptance ratio for liability set 3 = 0.000226
Acceptance ratio for liability set 4 = 0.000313
MCMC iteration = 1000
Acceptance ratio for liability set 1 = 0.102625
Acceptance ratio for liability set 2 = 0.380806
Acceptance ratio for liability set 3 = 0.307387
Acceptance ratio for liability set 4 = 0.115281
MCMC iteration = 2000
Acceptance ratio for liability set 1 = 0.156406
Acceptance ratio for liability set 2 = 0.425194
Acceptance ratio for liability set 3 = 0.356710
Acceptance ratio for liability set 4 = 0.174469
MCMC iteration = 3000
Acceptance ratio for liability set 1 = 0.168875
Acceptance ratio for liability set 2 = 0.420645
Acceptance ratio for liability set 3 = 0.353129
Acceptance ratio for liability set 4 = 0.194000
>
> # plotting log2(fold change) in response to heat stress for all genes
> HPDplot(model=mm,factors="conditionheat",main="response to heat stress")
>
>
>
>
>
>
> dev.off()
null device
1
>
|