Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
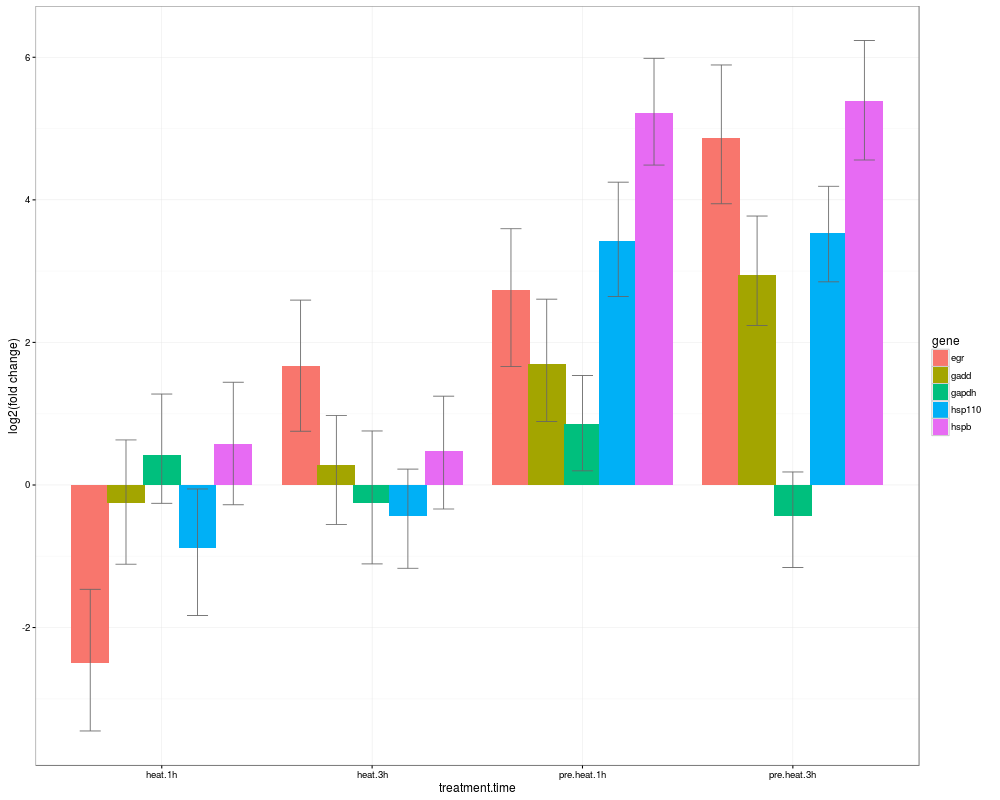
Summarizes and plots results of mcmc.qpcr function series.DescriptionCalculates abundances of each gene across factor combinations; calculates pairwise differences between all factor combinations and their significances for each gene; plots results as bar or line graphs with credible intervals (ggplot2) NOTE: only works for experiments involving a single multi-level fixed factor or two fully crossed multi-level fixed factors. UsageHPDsummary(model, data, xgroup=NULL,genes = NA, relative = FALSE, log.base = 2, summ.plot = TRUE, ptype="z", ...) Arguments
ValueA list of three items:
Author(s)Mikhail V. Matz, University of Texas at Austin <matz@utexas.edu> ReferencesMatz MV, Wright RM, Scott JG (2013) No Control Genes Required: Bayesian Analysis of qRT-PCR Data. PLoS ONE 8(8): e71448. doi:10.1371/journal.pone.0071448 See AlsoSee function summaryPlot() for plotting the summary table in other ways. Examplesdata(beckham.data) data(beckham.eff) # analysing the first 5 genes # (to try it with all 10 genes, change the line below to gcol=4:13) gcol=4:8 ccol=1:3 # columns containing experimental conditions # recalculating into molecule counts, reformatting qs=cq2counts(data=beckham.data,genecols=gcol, condcols=ccol,effic=beckham.eff,Cq1=37) # creating a single factor, 'treatment.time', out of 'tr' and 'time' qs$treatment.time=as.factor(paste(qs$tr,qs$time,sep=".")) # fitting a naive model naive=mcmc.qpcr( fixed="treatment.time", data=qs, nitt=3000,burnin=2000 # remove this line in actual analysis! ) #summary plot of inferred abundances # s1=HPDsummary(model=naive,data=qs) #summary plot of fold-changes relative to the global control s0=HPDsummary(model=naive,data=qs,relative=TRUE) # pairwise differences and their significances for each gene: s0$geneWise Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MCMC.qpcr)
Loading required package: MCMCglmm
Loading required package: Matrix
Loading required package: coda
Loading required package: ape
Loading required package: ggplot2
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/MCMC.qpcr/HPDsummary.Rd_%03d_medium.png", width=480, height=480)
> ### Name: HPDsummary
> ### Title: Summarizes and plots results of mcmc.qpcr function series.
> ### Aliases: HPDsummary
>
> ### ** Examples
>
>
> data(beckham.data)
> data(beckham.eff)
>
> # analysing the first 5 genes
> # (to try it with all 10 genes, change the line below to gcol=4:13)
> gcol=4:8
> ccol=1:3 # columns containing experimental conditions
>
> # recalculating into molecule counts, reformatting
> qs=cq2counts(data=beckham.data,genecols=gcol,
+ condcols=ccol,effic=beckham.eff,Cq1=37)
>
> # creating a single factor, 'treatment.time', out of 'tr' and 'time'
> qs$treatment.time=as.factor(paste(qs$tr,qs$time,sep="."))
>
> # fitting a naive model
> naive=mcmc.qpcr(
+ fixed="treatment.time",
+ data=qs,
+ nitt=3000,burnin=2000 # remove this line in actual analysis!
+ )
$PRIOR
$PRIOR$R
$PRIOR$R$V
[,1] [,2] [,3] [,4] [,5]
[1,] 1 0 0 0 0
[2,] 0 1 0 0 0
[3,] 0 0 1 0 0
[4,] 0 0 0 1 0
[5,] 0 0 0 0 1
$PRIOR$R$nu
[1] 4.002
$PRIOR$G
$PRIOR$G$G1
$PRIOR$G$G1$V
[1] 1
$PRIOR$G$G1$nu
[1] 0
$FIXED
[1] "count~0+gene++gene:treatment.time"
$RANDOM
[1] "~sample"
MCMC iteration = 0
Acceptance ratio for liability set 1 = 0.000385
Acceptance ratio for liability set 2 = 0.000167
Acceptance ratio for liability set 3 = 0.000143
Acceptance ratio for liability set 4 = 0.000450
Acceptance ratio for liability set 5 = 0.000462
MCMC iteration = 1000
Acceptance ratio for liability set 1 = 0.122667
Acceptance ratio for liability set 2 = 0.035238
Acceptance ratio for liability set 3 = 0.008357
Acceptance ratio for liability set 4 = 0.025250
Acceptance ratio for liability set 5 = 0.032821
MCMC iteration = 2000
Acceptance ratio for liability set 1 = 0.184846
Acceptance ratio for liability set 2 = 0.057357
Acceptance ratio for liability set 3 = 0.006000
Acceptance ratio for liability set 4 = 0.037975
Acceptance ratio for liability set 5 = 0.051436
MCMC iteration = 3000
Acceptance ratio for liability set 1 = 0.201769
Acceptance ratio for liability set 2 = 0.064619
Acceptance ratio for liability set 3 = 0.006452
Acceptance ratio for liability set 4 = 0.041425
Acceptance ratio for liability set 5 = 0.058154
>
> #summary plot of inferred abundances
> # s1=HPDsummary(model=naive,data=qs)
>
> #summary plot of fold-changes relative to the global control
> s0=HPDsummary(model=naive,data=qs,relative=TRUE)
>
> # pairwise differences and their significances for each gene:
> s0$geneWise
$egr
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -2.327858e+00 1.854407e+00 2.833691585 4.957891
heat.1h 4.483250e-05 NA 4.182265e+00 5.161549620 7.285749
heat.3h 2.281443e-03 2.926881e-11 NA 0.979284274 3.103483
pre.heat.1h 5.523180e-06 1.554312e-15 1.185160e-01 NA 2.124199
pre.heat.3h 4.440892e-16 0.000000e+00 5.571835e-07 0.001090859 NA
$gadd
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -6.020411e-02 3.237088e-01 1.78375467 3.010255
heat.1h 8.922235e-01 NA 3.839129e-01 1.84395878 3.070459
heat.3h 4.788135e-01 4.370183e-01 NA 1.46004590 2.686547
pre.heat.1h 3.470603e-05 1.349279e-04 9.962803e-04 NA 1.226501
pre.heat.3h 1.390690e-08 1.225225e-08 4.316809e-07 0.02109475 NA
$gapdh
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA 0.53610325 -0.13107656 0.96882842 -0.3005155
heat.1h 0.25878269 NA -0.66717981 0.43272517 -0.8366187
heat.3h 0.80256669 0.18697540 NA 1.09990498 -0.1694389
pre.heat.1h 0.04614115 0.38561023 0.02893827 NA -1.2693439
pre.heat.3h 0.54869907 0.09934648 0.73028210 0.01586384 NA
$hsp110
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -0.6720907 -3.867444e-01 3.5366114 3.5657399
heat.1h 1.733211e-01 NA 2.853463e-01 4.2087021 4.2378306
heat.3h 4.526113e-01 0.5877312 NA 3.9233558 3.9524843
pre.heat.1h 1.028644e-11 0.0000000 1.481038e-12 NA 0.0291285
pre.heat.3h 6.534062e-11 0.0000000 4.543921e-12 0.9593276 NA
$hspb
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA 0.6792636 4.657386e-01 5.3644282 5.4736501
heat.1h 0.1160298 NA -2.135250e-01 4.6851646 4.7943865
heat.3h 0.4060847 0.7056827 NA 4.8986896 5.0079115
pre.heat.1h 0.0000000 0.0000000 2.220446e-16 NA 0.1092219
pre.heat.3h 0.0000000 0.0000000 0.000000e+00 0.8411291 NA
>
>
>
>
>
>
> dev.off()
null device
1
>
|