Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Extracts qPCR model predictionsDescriptionGenerates a table of model-derived log2-transformed transcript abundances without global sample effects (i.e., corresponding to efficiency-corrected and normalized qPCR data) UsagegetNormalizedData(model, data, controls=NULL) Arguments
ValueThe function returns a list of two data frames. The first one, normData, is the model-predicted log2-transformed transcript abundances table. It has one column per gene and one row per sample. The second data frame, conditions, is a table of experimental conditions corresponding to the normData table. Author(s)Mikhail V. Matz, University of Texas at Austin <matz@utexas.edu> ReferencesMatz MV, Wright RM, Scott JG (2013) No Control Genes Required: Bayesian Analysis of qRT-PCR Data. PLoS ONE 8(8): e71448. doi:10.1371/journal.pone.0071448 Examples
library(MCMC.qpcr)
# loading Cq data and amplification efficiencies
data(coral.stress)
data(amp.eff)
genecolumns=c(5,6,16,17) # specifying columns corresponding to genes of interest
conditions=c(1:4) # specifying columns containing factors
# calculating molecule counts and reformatting:
dd=cq2counts(data=coral.stress,genecols=genecolumns,
condcols=conditions,effic=amp.eff,Cq1=37)
# fitting the model (must include random="sample", pr=TRUE options)
mm=mcmc.qpcr(
fixed="condition",
data=dd,
controls=c("nd5","rpl11"),
nitt=4000,
pr=TRUE,
random="sample"
)
# extracting model predictions
pp=getNormalizedData(mm,dd)
# here is the normalized data:
pp$normData
# and here are the corresponding conditions:
pp$conditions
# putting them together for plotting:
ppcombo=cbind(stack(pp$normData),rep(pp$conditions))
names(ppcombo)[1:2]=c("expression","gene")
# plotting boxplots of normalized data:
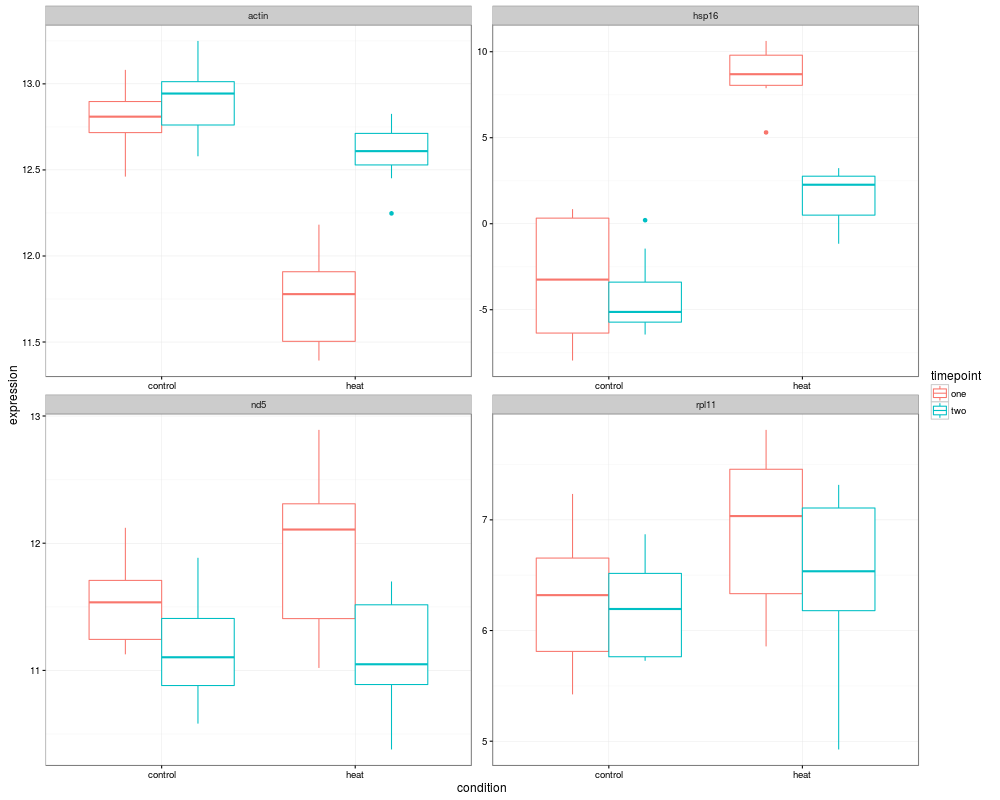
ggplot(ppcombo,aes(condition,expression,colour=timepoint))+
geom_boxplot()+
facet_wrap(~gene,scales="free")+
theme_bw()
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MCMC.qpcr)
Loading required package: MCMCglmm
Loading required package: Matrix
Loading required package: coda
Loading required package: ape
Loading required package: ggplot2
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/MCMC.qpcr/getNormalizedData.Rd_%03d_medium.png", width=480, height=480)
> ### Name: getNormalizedData
> ### Title: Extracts qPCR model predictions
> ### Aliases: getNormalizedData
>
> ### ** Examples
>
> library(MCMC.qpcr)
>
> # loading Cq data and amplification efficiencies
> data(coral.stress)
> data(amp.eff)
>
> genecolumns=c(5,6,16,17) # specifying columns corresponding to genes of interest
> conditions=c(1:4) # specifying columns containing factors
>
> # calculating molecule counts and reformatting:
> dd=cq2counts(data=coral.stress,genecols=genecolumns,
+ condcols=conditions,effic=amp.eff,Cq1=37)
>
> # fitting the model (must include random="sample", pr=TRUE options)
> mm=mcmc.qpcr(
+ fixed="condition",
+ data=dd,
+ controls=c("nd5","rpl11"),
+ nitt=4000,
+ pr=TRUE,
+ random="sample"
+ )
$PRIOR
$PRIOR$B
$PRIOR$B$mu
[1] 0 0 0 0 0 0 0 0
$PRIOR$B$V
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
[1,] 1e+08 0e+00 0e+00 0e+00 0e+00 0e+00 0.0000000 0.0000000
[2,] 0e+00 1e+08 0e+00 0e+00 0e+00 0e+00 0.0000000 0.0000000
[3,] 0e+00 0e+00 1e+08 0e+00 0e+00 0e+00 0.0000000 0.0000000
[4,] 0e+00 0e+00 0e+00 1e+08 0e+00 0e+00 0.0000000 0.0000000
[5,] 0e+00 0e+00 0e+00 0e+00 1e+08 0e+00 0.0000000 0.0000000
[6,] 0e+00 0e+00 0e+00 0e+00 0e+00 1e+08 0.0000000 0.0000000
[7,] 0e+00 0e+00 0e+00 0e+00 0e+00 0e+00 0.3663091 0.0000000
[8,] 0e+00 0e+00 0e+00 0e+00 0e+00 0e+00 0.0000000 0.3663091
$PRIOR$R
$PRIOR$R$V
[,1] [,2] [,3] [,4]
[1,] 1 0 0 0
[2,] 0 1 0 0
[3,] 0 0 1 0
[4,] 0 0 0 1
$PRIOR$R$nu
[1] 3.002
$PRIOR$G
$PRIOR$G$G1
$PRIOR$G$G1$V
[1] 1
$PRIOR$G$G1$nu
[1] 0
$PRIOR$G$G2
$PRIOR$G$G2$V
[,1] [,2] [,3] [,4]
[1,] 1 0 0 0
[2,] 0 1 0 0
[3,] 0 0 1 0
[4,] 0 0 0 1
$PRIOR$G$G2$nu
[1] 0
$FIXED
[1] "count~0+gene++gene:condition"
$RANDOM
[1] "~sample+idh(gene):sample"
MCMC iteration = 0
Acceptance ratio for liability set 1 = 0.000266
Acceptance ratio for liability set 2 = 0.000635
Acceptance ratio for liability set 3 = 0.000365
Acceptance ratio for liability set 4 = 0.000203
MCMC iteration = 1000
Acceptance ratio for liability set 1 = 0.125984
Acceptance ratio for liability set 2 = 0.311238
Acceptance ratio for liability set 3 = 0.303825
Acceptance ratio for liability set 4 = 0.156094
MCMC iteration = 2000
Acceptance ratio for liability set 1 = 0.188672
Acceptance ratio for liability set 2 = 0.333730
Acceptance ratio for liability set 3 = 0.336190
Acceptance ratio for liability set 4 = 0.225453
MCMC iteration = 3000
Acceptance ratio for liability set 1 = 0.221359
Acceptance ratio for liability set 2 = 0.337667
Acceptance ratio for liability set 3 = 0.339937
Acceptance ratio for liability set 4 = 0.248109
MCMC iteration = 4000
Acceptance ratio for liability set 1 = 0.240172
Acceptance ratio for liability set 2 = 0.318571
Acceptance ratio for liability set 3 = 0.337587
Acceptance ratio for liability set 4 = 0.264609
>
> # extracting model predictions
> pp=getNormalizedData(mm,dd)
>
> # here is the normalized data:
> pp$normData
hsp16 actin nd5 rpl11
1 -3.47795277 12.87418 11.28497 7.297597
2 8.02000728 12.12638 10.94048 7.733618
3 -3.19970389 12.71495 12.09723 7.190185
4 8.16632198 12.10252 11.25693 7.123951
5 1.19866768 12.99997 12.27613 5.611415
6 10.79611433 11.68718 12.52507 7.802915
7 -3.43122561 13.15043 11.76310 5.637348
8 7.84303179 11.97914 12.21349 7.527204
9 -5.84196734 12.89903 11.58675 6.606744
10 9.92617984 11.78224 13.17928 6.552712
11 -5.48344313 13.07968 11.54714 5.929344
12 5.41325748 11.94335 12.49211 6.398555
13 0.73702153 12.80468 11.22311 6.275081
14 10.10344639 12.10784 11.56307 5.954321
15 0.35690004 12.98063 11.38833 6.650236
16 9.32905374 11.80912 12.27635 7.319336
17 -7.87154891 13.16756 10.41543 6.645269
18 3.02292658 12.51073 10.63188 7.074402
19 -5.00036847 13.08979 11.06974 6.448193
20 2.92466513 12.41626 10.81831 6.977519
21 -0.01102747 13.05266 11.52856 5.736194
22 2.67675379 12.17872 11.45970 6.604527
23 -0.98393605 13.00210 10.83387 7.054781
24 0.29151508 12.51993 10.13565 7.090342
25 -3.34059262 12.82451 11.42666 6.350209
26 0.23181204 12.35630 11.66513 4.756375
27 -5.34260503 12.81584 11.94863 6.034343
28 1.79189767 12.36196 11.45611 6.134166
29 -5.91553808 13.04340 10.93324 6.522172
30 2.50818704 12.46471 10.89419 6.386004
31 -4.56205184 12.96699 11.61048 5.931506
32 -0.55396734 12.51844 10.87108 6.130582
>
> # and here are the corresponding conditions:
> pp$conditions
individual condition timepoint
1 s1 control one
2 s1 heat one
3 s2 control one
4 s2 heat one
5 s4 control one
6 s4 heat one
7 s5 control one
8 s5 heat one
9 s8 control one
10 s8 heat one
11 s12 control one
12 s12 heat one
13 s13 control one
14 s13 heat one
15 s15 control one
16 s15 heat one
17 s1 control two
18 s1 heat two
19 s2 control two
20 s2 heat two
21 s4 control two
22 s4 heat two
23 s5 control two
24 s5 heat two
25 s8 control two
26 s8 heat two
27 s12 control two
28 s12 heat two
29 s13 control two
30 s13 heat two
31 s15 control two
32 s15 heat two
>
> # putting them together for plotting:
> ppcombo=cbind(stack(pp$normData),rep(pp$conditions))
> names(ppcombo)[1:2]=c("expression","gene")
>
> # plotting boxplots of normalized data:
> ggplot(ppcombo,aes(condition,expression,colour=timepoint))+
+ geom_boxplot()+
+ facet_wrap(~gene,scales="free")+
+ theme_bw()
>
>
>
>
>
> dev.off()
null device
1
>
|