Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Analyzes qRT-PCR data using generalized linear mixed modelDescriptionWrapper function for MCMCglmm by Jarrod Hadfield, designed for qRT-PCR data analysis. Usagemcmc.qpcr(fixed=NULL, globalFixed=NULL, random = NULL, globalRandom=NULL, data, controls = NULL, normalize=FALSE, include = NULL, m.fix = 1.2, v.fix = NULL, geneSpecRes=TRUE, Covar=FALSE, vprior="uninf", ...) Arguments
DetailsThis function constructs priors and runs an MCMC chain to fit a Poisson-lognormal generalized linear mixed model to the counts data. The fixed effects for the model by default assume a gene-specific intercept, and gene-specific effect for each of the listed fixed factors. The user-specified random effects are all assumed to be gene-specific with no covariances. There is also a universal random factor: the scalar random effect of sample, which accounts for the unequal template loading. Residual variances are assumed to be gene-specific with no covariances, with weakly informative inverse Wishart prior (variance=1, nu=[number of genes]-0.998). The priors for fixed effects are diffuse gaussians with a mean at 0 and very large variances (1e+8), except for control genes, for which the prior variances are defined by the m.fix parameter. For the gene-specific random effects and residual variation, non-informative priors are used to achieve results equivalent to ML estimation. For control genes, when v.fix parameter is specified, it will be used to restrict residial variance. Both m.fix and v.fix parameters should be specified as allowed average fold-change, so the lowest they can go is 1 (no variation). All control genes share the same m.fix and v.fix parameters. ValueAn MCMCglmm object. HPDsummary() function within this package summaizes these data, calculates all gene-wise credible intervals and p-values, and plots the results either as line-point-whiskers graph or a bar-whiskers graph using ggplot2 functions. HPDsummary() only works for experiments with a single multilevel factor or two fully crossed multilevel factors. Use finctions HPDplot(), HPDplotBygene() and HPDplotBygeneBygroup() to summarize and plot more complicated designs. For more useful operations on MCMCglmm objects, such as posterior.mode(), HPDinterval(), and plot(), see documentation for MCMCglmm package. Author(s)Mikhail V. Matz, University of Texas at Austin <matz@utexas.edu> ReferencesMatz MV, Wright RM, Scott JG (2013) No Control Genes Required: Bayesian Analysis of qRT-PCR Data. PLoS ONE 8(8): e71448. doi:10.1371/journal.pone.0071448 Examplesdata(beckham.data) data(beckham.eff) # analysing the first 5 genes # (to try it with all 10 genes, change the line below to gcol=4:13) gcol=4:8 ccol=1:3 # columns containing experimental conditions # recalculating into molecule counts, reformatting qs=cq2counts(data=beckham.data,genecols=gcol, condcols=ccol,effic=beckham.eff,Cq1=37) # creating a single factor, 'treatment.time', out of 'tr' and 'time' qs$treatment.time=as.factor(paste(qs$tr,qs$time,sep=".")) # fitting a naive model naive=mcmc.qpcr( fixed="treatment.time", data=qs, nitt=3000,burnin=2000 # remove this line in actual analysis! ) #summary plot of inferred abundances #s1=HPDsummary(model=naive,data=qs) #summary plot of fold-changes relative to the global control s0=HPDsummary(model=naive,data=qs,relative=TRUE) # pairwise differences and their significances for each gene: s0$geneWise Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MCMC.qpcr)
Loading required package: MCMCglmm
Loading required package: Matrix
Loading required package: coda
Loading required package: ape
Loading required package: ggplot2
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/MCMC.qpcr/mcmc.qpcr.Rd_%03d_medium.png", width=480, height=480)
> ### Name: mcmc.qpcr
> ### Title: Analyzes qRT-PCR data using generalized linear mixed model
> ### Aliases: mcmc.qpcr
>
> ### ** Examples
>
>
> data(beckham.data)
> data(beckham.eff)
>
> # analysing the first 5 genes
> # (to try it with all 10 genes, change the line below to gcol=4:13)
> gcol=4:8
> ccol=1:3 # columns containing experimental conditions
>
> # recalculating into molecule counts, reformatting
> qs=cq2counts(data=beckham.data,genecols=gcol,
+ condcols=ccol,effic=beckham.eff,Cq1=37)
>
> # creating a single factor, 'treatment.time', out of 'tr' and 'time'
> qs$treatment.time=as.factor(paste(qs$tr,qs$time,sep="."))
>
> # fitting a naive model
> naive=mcmc.qpcr(
+ fixed="treatment.time",
+ data=qs,
+ nitt=3000,burnin=2000 # remove this line in actual analysis!
+ )
$PRIOR
$PRIOR$R
$PRIOR$R$V
[,1] [,2] [,3] [,4] [,5]
[1,] 1 0 0 0 0
[2,] 0 1 0 0 0
[3,] 0 0 1 0 0
[4,] 0 0 0 1 0
[5,] 0 0 0 0 1
$PRIOR$R$nu
[1] 4.002
$PRIOR$G
$PRIOR$G$G1
$PRIOR$G$G1$V
[1] 1
$PRIOR$G$G1$nu
[1] 0
$FIXED
[1] "count~0+gene++gene:treatment.time"
$RANDOM
[1] "~sample"
MCMC iteration = 0
Acceptance ratio for liability set 1 = 0.000410
Acceptance ratio for liability set 2 = 0.000262
Acceptance ratio for liability set 3 = 0.000286
Acceptance ratio for liability set 4 = 0.000350
Acceptance ratio for liability set 5 = 0.000436
MCMC iteration = 1000
Acceptance ratio for liability set 1 = 0.121692
Acceptance ratio for liability set 2 = 0.037310
Acceptance ratio for liability set 3 = 0.009333
Acceptance ratio for liability set 4 = 0.025975
Acceptance ratio for liability set 5 = 0.031256
MCMC iteration = 2000
Acceptance ratio for liability set 1 = 0.181615
Acceptance ratio for liability set 2 = 0.056786
Acceptance ratio for liability set 3 = 0.005429
Acceptance ratio for liability set 4 = 0.038100
Acceptance ratio for liability set 5 = 0.047974
MCMC iteration = 3000
Acceptance ratio for liability set 1 = 0.201231
Acceptance ratio for liability set 2 = 0.065238
Acceptance ratio for liability set 3 = 0.006357
Acceptance ratio for liability set 4 = 0.044175
Acceptance ratio for liability set 5 = 0.057077
>
> #summary plot of inferred abundances
> #s1=HPDsummary(model=naive,data=qs)
>
> #summary plot of fold-changes relative to the global control
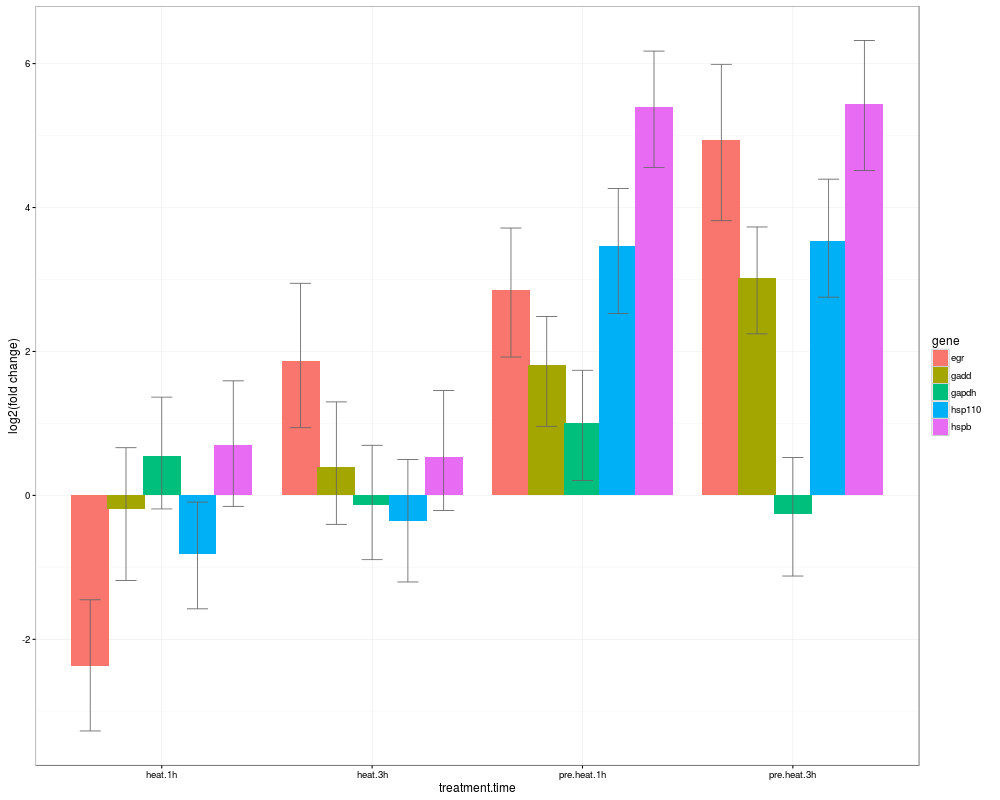
> s0=HPDsummary(model=naive,data=qs,relative=TRUE)
>
> # pairwise differences and their significances for each gene:
> s0$geneWise
$egr
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -2.537170e+00 1.736627e+00 2.788052747 4.784214
heat.1h 5.524789e-05 NA 4.273797e+00 5.325222705 7.321384
heat.3h 6.857999e-03 1.863398e-11 NA 1.051426065 3.047587
pre.heat.1h 7.172600e-07 0.000000e+00 8.746079e-02 NA 1.996161
pre.heat.3h 1.228173e-11 0.000000e+00 6.997938e-06 0.001912884 NA
$gadd
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -2.617963e-01 2.705140e-01 1.7725979 2.939627
heat.1h 6.415405e-01 NA 5.323103e-01 2.0343941 3.201424
heat.3h 5.771007e-01 3.190757e-01 NA 1.5020838 2.669113
pre.heat.1h 5.625912e-04 4.714488e-05 1.662368e-03 NA 1.167030
pre.heat.3h 1.191377e-09 3.857357e-10 7.774618e-08 0.0261633 NA
$gapdh
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA 0.5204458 -0.21179205 0.939611747 -0.3210171
heat.1h 0.32440560 NA -0.73223784 0.419165953 -0.8414629
heat.3h 0.66530974 0.1570238 NA 1.151403793 -0.1092250
pre.heat.1h 0.05897014 0.3485469 0.01276365 NA -1.2606288
pre.heat.3h 0.48572579 0.0910167 0.81388843 0.008308123 NA
$hsp110
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA -0.8647825 -4.851809e-01 3.4637608 3.44729725
heat.1h 8.684040e-02 NA 3.796015e-01 4.3285433 4.31207970
heat.3h 3.940509e-01 0.5106292 NA 3.9489417 3.93247816
pre.heat.1h 1.558531e-12 0.0000000 2.997602e-14 NA -0.01646355
pre.heat.3h 1.150628e-10 0.0000000 1.234191e-11 0.9751703 NA
$hspb
difference
pvalue control.0h heat.1h heat.3h pre.heat.1h pre.heat.3h
control.0h NA 0.6014920 0.4917521 5.3284417 5.4289563
heat.1h 0.1948475 NA -0.1097400 4.7269496 4.8274643
heat.3h 0.3499879 0.8334443 NA 4.8366896 4.9372042
pre.heat.1h 0.0000000 0.0000000 0.0000000 NA 0.1005147
pre.heat.3h 0.0000000 0.0000000 0.0000000 0.8660549 NA
>
>
>
>
>
>
> dev.off()
null device
1
>
|