Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Class mergeExpressionSet, a class for merged microarray data, and methods for processing themDescriptionThis is class representation for merged Microarray Data. DetailsThe mergeExpressionSet class is conceived as an extension of the ExpressionSet class provided in Biobase for the storage of expression array data. A mergeExpressionSet object is primarily a list of ExpressionSet objects, along with an incidence matrix indicating which genes appear in which studies. A mergeExpressionSet object with a single study reverts to the ExpressionSet class. A number of accessor functions are defiined for this class, as well as a few convenient analysis and plotting functions. SlotsWe assume there are K studies, representing a total of M unique genes.
MethodsDerived from
Class-specific methods:
Standard generic methods:
See Also
Examples
if(require(Biobase) & require(MASS)){
data(mergeData)
merged <-mergeExprs(sample1,sample2,sample3)
merged[1:2]
i<-c(1,3)
merged[i]
exprs(merged)
names(merged)<-c("study1","study2","study3")
length(merged)
summary(merged)
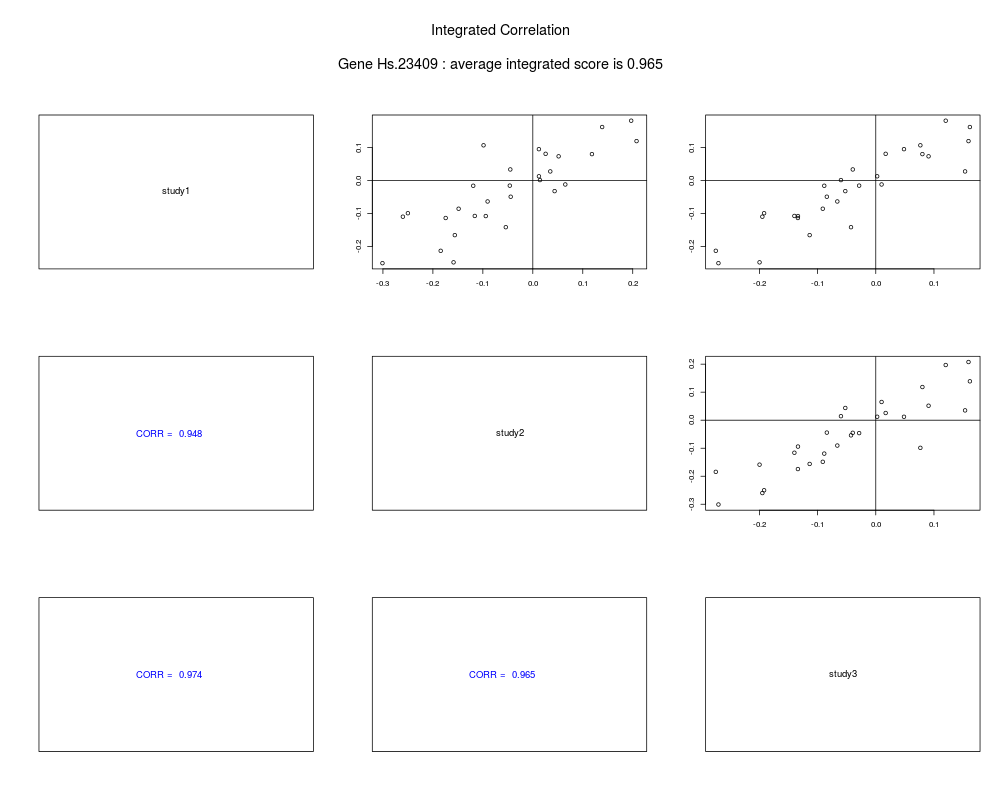
plot(merged)
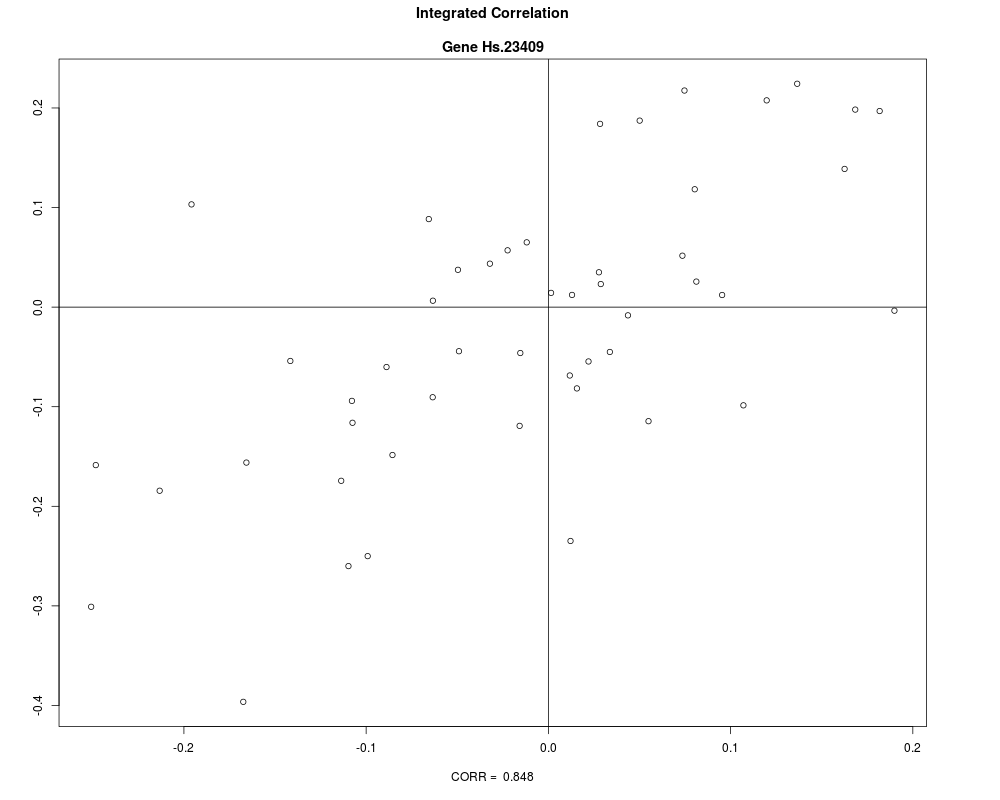
plot(merged[1:2])
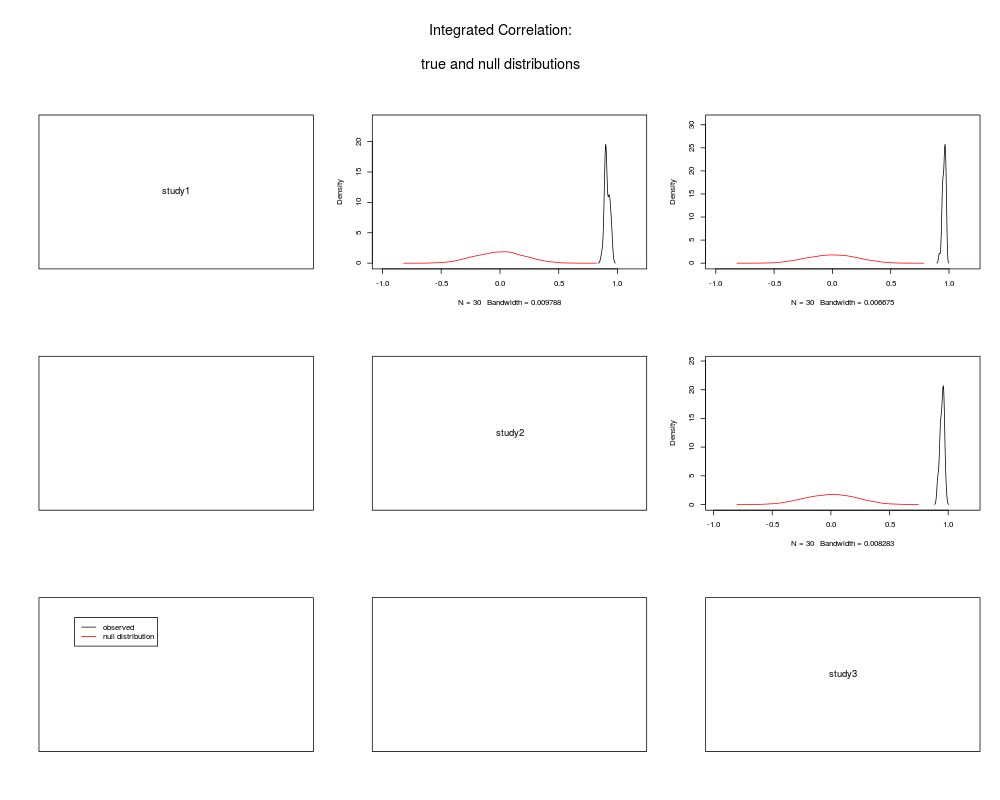
intcorDens(merged)
inter <- intersection(merged)
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MergeMaid)
Loading required package: survival
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: MASS
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/MergeMaid/class.mergeExpressionSet.Rd_%03d_medium.png", width=480, height=480)
> ### Name: mergeExpressionSet
> ### Title: Class mergeExpressionSet, a class for merged microarray data,
> ### and methods for processing them
> ### Aliases: mergeExpressionSet-class mergeExpressionSet geneNames
> ### geneNames<- geneStudy geneStudy<- subsetmES
> ### geneStudy,mergeExpressionSet-method names,mergeExpressionSet-method
> ### length,mergeExpressionSet-method summary,mergeExpressionSet-method
> ### show,mergeExpressionSet-method exprs<-,mergeExpressionSet,ANY-method
> ### exprs,mergeExpressionSet-method geneNames<-,mergeExpressionSet-method
> ### geneNames,mergeExpressionSet-method
> ### notes<-,mergeExpressionSet,ANY-method notes,mergeExpressionSet-method
> ### phenoData<-,mergeExpressionSet,ANY-method
> ### phenoData,mergeExpressionSet-method names<-,mergeExpressionSet-method
> ### geneStudy<-,mergeExpressionSet-method [,mergeExpressionSet-method
> ### plot,mergeExpressionSet-method intcorDens,mergeExpressionSet-method
> ### intCor,mergeExpressionSet-method
> ### intersection,mergeExpressionSet-method
> ### modelOutcome,mergeExpressionSet-method isna maxintcor
> ### Keywords: classes
>
> ### ** Examples
>
> if(require(Biobase) & require(MASS)){
+ data(mergeData)
+ merged <-mergeExprs(sample1,sample2,sample3)
+
+ merged[1:2]
+ i<-c(1,3)
+ merged[i]
+
+ exprs(merged)
+
+ names(merged)<-c("study1","study2","study3")
+
+ length(merged)
+
+ summary(merged)
+
+ plot(merged)
+
+ plot(merged[1:2])
+
+ intcorDens(merged)
+
+ inter <- intersection(merged)
+ }
>
>
>
>
>
> dev.off()
null device
1
>
|