Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
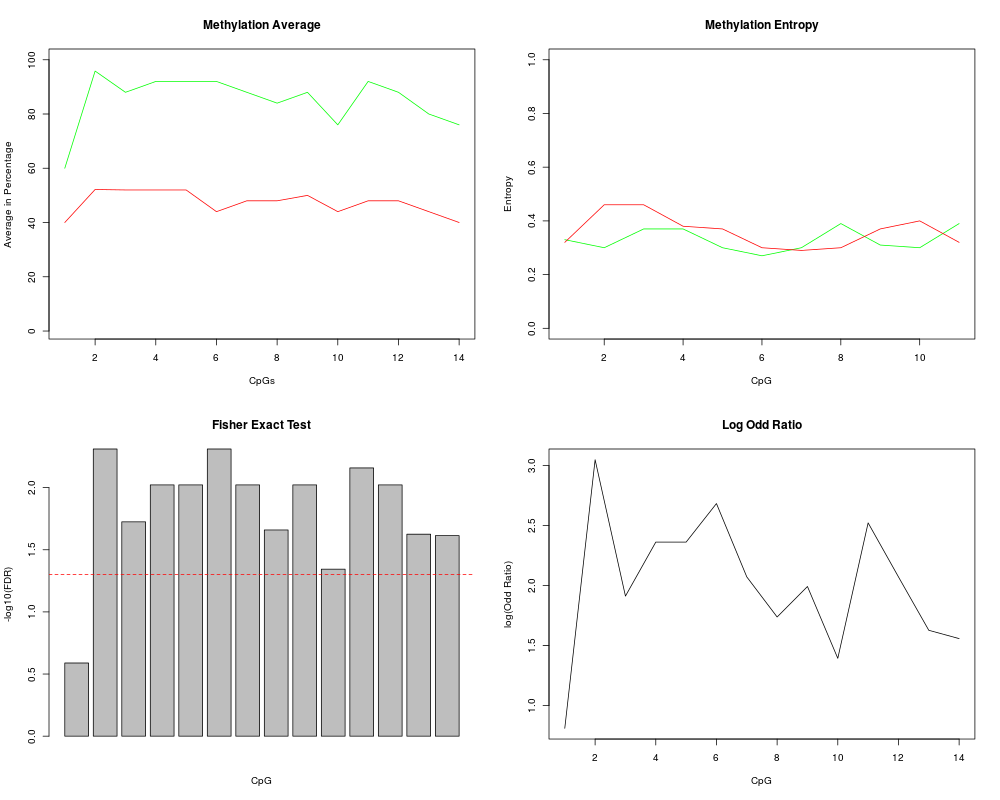
Complete Methylation Analysis of Next Generation Sequencing DataDescriptionThis function perform complete methylation analysis of the data. 1. Visualize methylation pattern 2. Calculate methylation average 3. Calculate methylation entropy 4. Perform fisher exact test on the samples to identify significant CpG sites. Usagecompare_samples(healthy, tumor) Arguments
ValueGenerate a plot of Methylation Average, Methylation Entropy, Fisher Exact Test and Log Odd Ratio NoteThis function needs time to process depending on the number of sequences in fasta file Author(s)Muhammad Ahmer Jamil, Prof. Holger Frohlich, Priv.-Doz. Dr. Osman El-Maarri Maintainer: Muhammad Ahmer Jamil engr.ahmerjamil@gmail.com See Also
Examples
healthy = system.file("extdata", "Healthy.fasta", package = "MethTargetedNGS")
tumor = system.file("extdata", "Tumor.fasta", package = "MethTargetedNGS")
reference = system.file("extdata", "Reference.fasta", package = "MethTargetedNGS")
healthy = methAlign(healthy,reference)
tumor = methAlign(tumor,reference)
compare_samples(healthy,tumor)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(MethTargetedNGS)
Loading required package: stringr
Loading required package: seqinr
Loading required package: gplots
Attaching package: 'gplots'
The following object is masked from 'package:stats':
lowess
Loading required package: Biostrings
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following object is masked from 'package:gplots':
space
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Attaching package: 'Biostrings'
The following object is masked from 'package:seqinr':
translate
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/MethTargetedNGS/compare_samples.Rd_%03d_medium.png", width=480, height=480)
> ### Name: compare_samples
> ### Title: Complete Methylation Analysis of Next Generation Sequencing Data
> ### Aliases: compare_samples
> ### Keywords: Methylation Average Entropy Odd Ratio Significant CpG sites
> ### Fisher Test
>
> ### ** Examples
>
> healthy = system.file("extdata", "Healthy.fasta", package = "MethTargetedNGS")
> tumor = system.file("extdata", "Tumor.fasta", package = "MethTargetedNGS")
> reference = system.file("extdata", "Reference.fasta", package = "MethTargetedNGS")
>
> healthy = methAlign(healthy,reference)
Time difference of 2.25 secs
> tumor = methAlign(tumor,reference)
Time difference of 2.18 secs
> compare_samples(healthy,tumor)
Time difference of 0.02 secs
Time difference of 0.02 secs
>
>
>
>
>
> dev.off()
null device
1
>
|
Created & Maintained by Osamu Ogasawara (osamu.ogasawara@gmail.com) and 