Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Between-array scalingDescriptionThis function performs an between-array scaling Usagebas(obj,mode="var") Arguments
DetailsThe function Following schemes (
NoteBetween-array scaling should only be performed if it can be assumed that the different arrays have a similar distribution of logged ratios. This has to be check on a case-by-case basis. Caution should be taken in the interpretation of results for arrays hybridised with biologically divergent samples, if between-array scaling is applied. Author(s)Matthias E. Futschik (http://itb.biologie.hu-berlin.de/~futschik) ReferencesBolstad et al., A comparison of normalization methods for high density oligonucleotide array data based on variance and bias, Bioinformatics, 19: 185-193, 2003 See Also
Examples
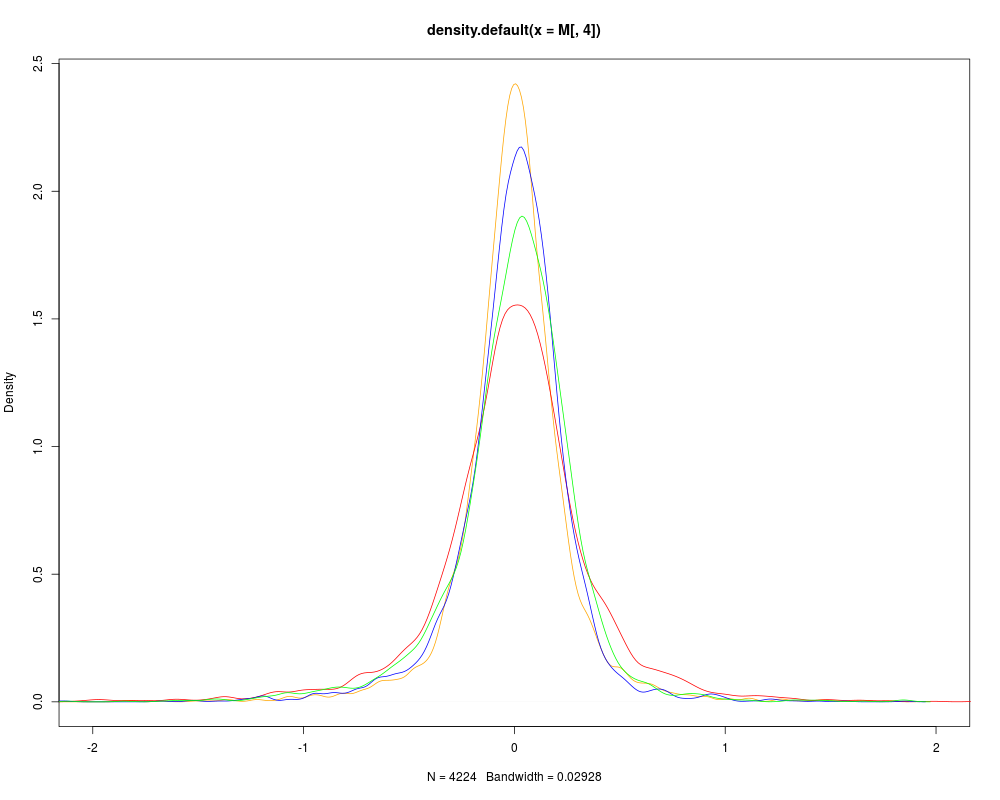
# DISTRIBUTION OF M BEFORE SCALING
data(sw.olin)
col <- c("red","blue","green","orange")
M <- maM(sw.olin)
plot(density(M[,4]),col=col[4],xlim=c(-2,2))
for (i in 1:3){
lines(density(M[,i]),col=col[i])
}
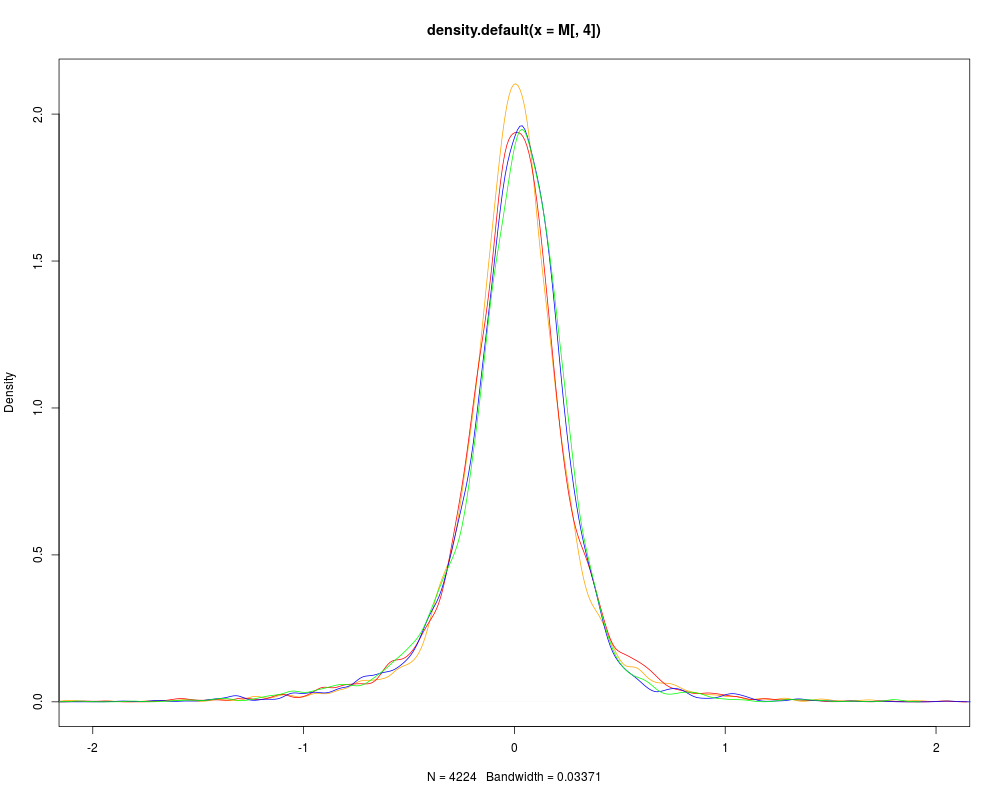
# SCALING AND VISUALISATION
sw.olin.s <- bas(sw.olin,mode="var")
M <- maM(sw.olin.s)
plot(density(M[,4]),col=col[4],xlim=c(-2,2))
for (i in 1:3){
lines(density(M[,i]),col=col[i])
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(OLIN)
Loading required package: locfit
locfit 1.5-9.1 2013-03-22
Loading required package: marray
Loading required package: limma
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/OLIN/bas.Rd_%03d_medium.png", width=480, height=480)
> ### Name: bas
> ### Title: Between-array scaling
> ### Aliases: bas
> ### Keywords: utilities
>
> ### ** Examples
>
>
>
> # DISTRIBUTION OF M BEFORE SCALING
> data(sw.olin)
>
> col <- c("red","blue","green","orange")
> M <- maM(sw.olin)
>
> plot(density(M[,4]),col=col[4],xlim=c(-2,2))
> for (i in 1:3){
+ lines(density(M[,i]),col=col[i])
+ }
>
>
> # SCALING AND VISUALISATION
> sw.olin.s <- bas(sw.olin,mode="var")
>
> M <- maM(sw.olin.s)
>
> plot(density(M[,4]),col=col[4],xlim=c(-2,2))
> for (i in 1:3){
+ lines(density(M[,i]),col=col[i])
+ }
>
>
>
>
>
>
> dev.off()
null device
1
>
|