R: The 'Proteins' Class for Proteomics Data And Meta-Data
Proteins-class
R Documentation
The Proteins Class for Proteomics Data And Meta-Data
Description
The Proteins class encapsulates data and meta-data for
proteomics experiments. The class stores the protein sequences as well
as specific subsets of interest, typically peptides, as ranges. The
Proteins instances, the sequence and peptide slots are
described by their respective metadata attributes.
Objects from the Class
Objects can be created using its constructor Proteins. The
constructor either takes a fasta file name as first argument
or, alternatively a named uniprotIds argument with valid
UniProt accession numbers (not yet implemented).
Details
An instance of class Proteins is characterised by one or
multiple protein sequences that can be accessed as AAStringSet
with the aa accessor. Sequence-specific annotation, such as
accession numbers, protein and gene names, ... is available with the
ametadata or acols methods. General metadata such as the
data of creation of the instance are stored as a list returned
by the metadata accessor, which would typically contain a
created character that documents when the object was created, a
reference genome descriptor, a UniProtRelease with the
release data of the UniProt database and possibly others.
Each sequence of a Proteins instance can also be characterised
by a set of specific ranges describing peptides of interest. These
peptide features can be extracted as an AAStringSetList,
where each protein sequence contains 0 or more peptide features. These
peptides features are encode as ranges along the original proteins
sequences (a list of IRanges) that can be extracted with
the pranges function. These peptide features have their own
metadata describing for example peptide identification scores, number
of missed cleavages, ... available with the pmetadata or
pcols methods.
See also the Pbase-data vignette.
Developement notes
Since version 0.2.0, addIdentificationData supports multiple
identification file names to be added to a Proteins instance
(argument renamed filenames) using either mzID or
mzR. Added new Pparams parametrisation infrastructure.
See news(package = "Pbase") for a description of all changes.
Other possible metadata fields: Uniprot.sw, biomaRt
instances.
Slots
metadata:
Object of class "list" containing
global metadata, accessed with metadata.
aa:
Object of class "AAStringSet" storing the
protein sequences, accessed with aa.
pranges:
Object of class
"CompressedIRangesList" containg protein feature ranges
such as theoretical (obtain by in silico cleavage) or observed
peptides. Accessed as an IRangesList with
pranges or and AAStringSetList with
pfeatures.
.__classVersion__:
Object of class "Versions"
documenting the class verions. Intended for developer use and
debugging.
Extends
Class "Versioned", directly.
Methods
aa
signature(x = "Proteins"): Returns an
AAStringSet instance representing the
sequences of the proteins.
pfeatures
signature(x = "Proteins"): ...
pranges
signature(x = "Proteins"): ...
metadata
signature(x = "Proteins"): Returns a
list of global metadata of the instance x, including
data of instance creation or, if created from a set of UnitProt
identifiers (see constructors above), the UniProt version and
UnitProt.WS version number.
ametadata
signature(x = "Proteins"): Returns a
DataFrame of protein metadata.
acols
signature(x = "Proteins"): See
ametadata.
pmetadata
signature(x = "Proteins"): Returns a
list of feature metadata.
pcols
signature(x = "Proteins"): See
pmetadata.
avarLabels
signature(x = "Proteins"): Returns the
names of the sequences metadata.
pvarLabels
signature(x = "Proteins"): Returns the
names of the peptide feature metadata.
seqnames
signature(x = "Proteins"): Returns the
protein sequence names defined as UniProt accession numbers.
length
signature(x = "Proteins"): Returns the number
of proteins.
[
signature(x = "Proteins", i = "ANY", j = "missing"):
Creates a subset of the Proteins insance.
[[
signature(x = "Proteins", i = "ANY", j = "missing"):
Returns an AAString instance representing the
sequence of the selected protein.
pfilter
signature(x = "Proteins", mass = "numeric", len
= "numeric", ...): ...
cleave
signature(x = "Proteins", enzym = "character",
missedCleavages = "numeric"):
Cleaves all proteins using the enzym rule while allowing
missedCleavages missing cleavages. Please see
cleave for details.
addIdentificationData
signature(object = "Proteins",
id = "character", rmEmptyRanges = "logical", par =
"Pparams"): Adds identification data from an IdentMzMl file (id)
to the Proteins object. If rmEmptyRanges is TRUE
proteins without any identification data are removed. See
Pparams for further settings.
addPeptideFragments
signature(object = "Proteins",
filenames = "character", rmEmptyRanges = "logical", par =
"Pparams"): Adds identification data from a fasta file (filenames)
to the Proteins object. Please note that both fasta files (the origin
of the Proteins object and the ones given in filenames) must
share the same Uniprot accession numbers.
If rmEmptyRanges is TRUE
proteins without any identification data are removed. See
Pparams for further settings.
plot
signature(x = "Proteins", y = "missing"): Plots
all proteins and associated peptides using the
Gviz/Pviz infrastructure.
show
signature(object = "Proteins"): Displays object
summary as text.
Functions
rmEmptyRanges
signature(x = "Proteins")
: removes
proteins with empty peptide ranges.
proteotypic
signature(x = "Proteins"): returns a
modified Proteins object. pcols(x) gains a "Proteotypic"
logical column, indicating of the peptide is proteotypic or now.
proteinCoverage
signature(pattern = "Proteins"):
calulates the coverage of proteins. pcols(x) gains a
"Coverage" numeric column.
isCleaver
signature(x = "Proteins", missedCleavages =
"numeric"): Tests whether a Protein object was cleaved
already.
Author(s)
Laurent Gatto <lg390@cam.ac.uk> and Sebastian Gibb <mail@sebastiangibb.de>
## Create a Protein object reading all proteins from a fasta file.
fastaFiles <- list.files(system.file("extdata", package = "Pbase"),
pattern = "fasta", full.names = TRUE)
p <- Proteins(fastaFiles)
p
metadata(p)
## Adding custom metadata
metadata(p, "Comment") <- "I love R"
metadata(p)
## Plotting
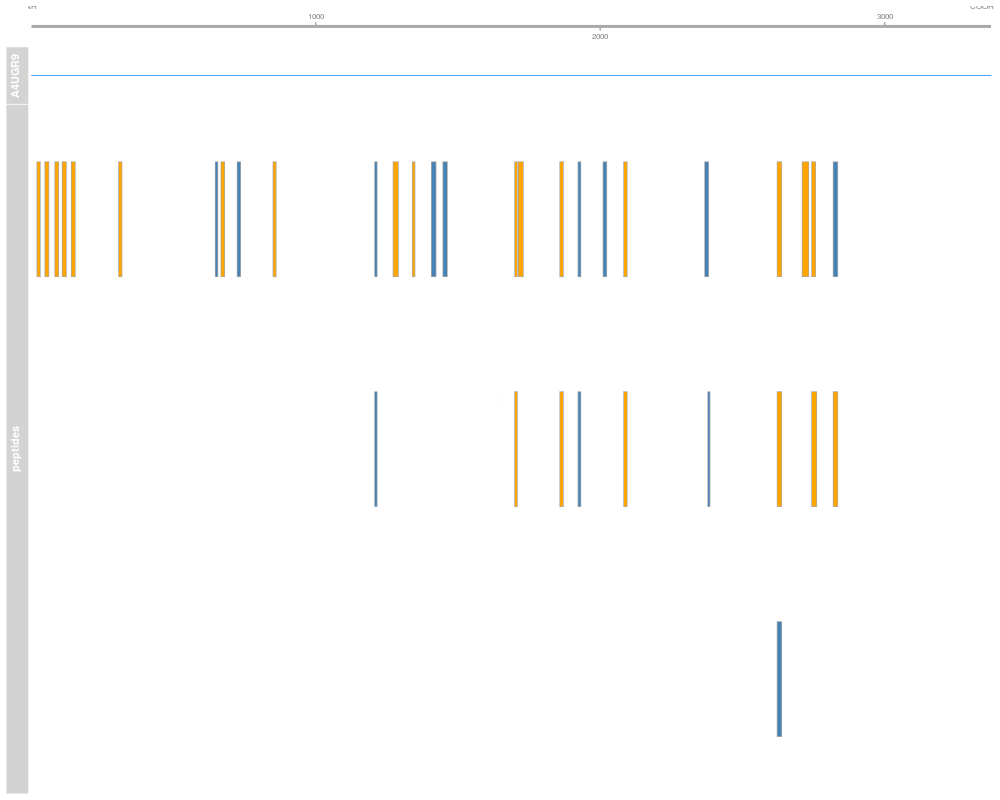
plot(p[1:5], from = 1, to = 30)
## Cleaving
pp <- cleave(p[1:100])
pp <- proteotypic(pp)
pp
pcols(pp[1:2])
plot(pp[1:2], from = 20, to = 30)
## Protein coverage
pp <- proteinCoverage(pp)
avarLabels(pp)
acols(pp)$Coverage
pp
## Add indentification data
idfile <- system.file("extdata/Thermo_Hela_PRTC_selected.mzid",
package = "Pbase")
p <- addIdentificationData(p, idfile)
pranges(p)
pfeatures(p)
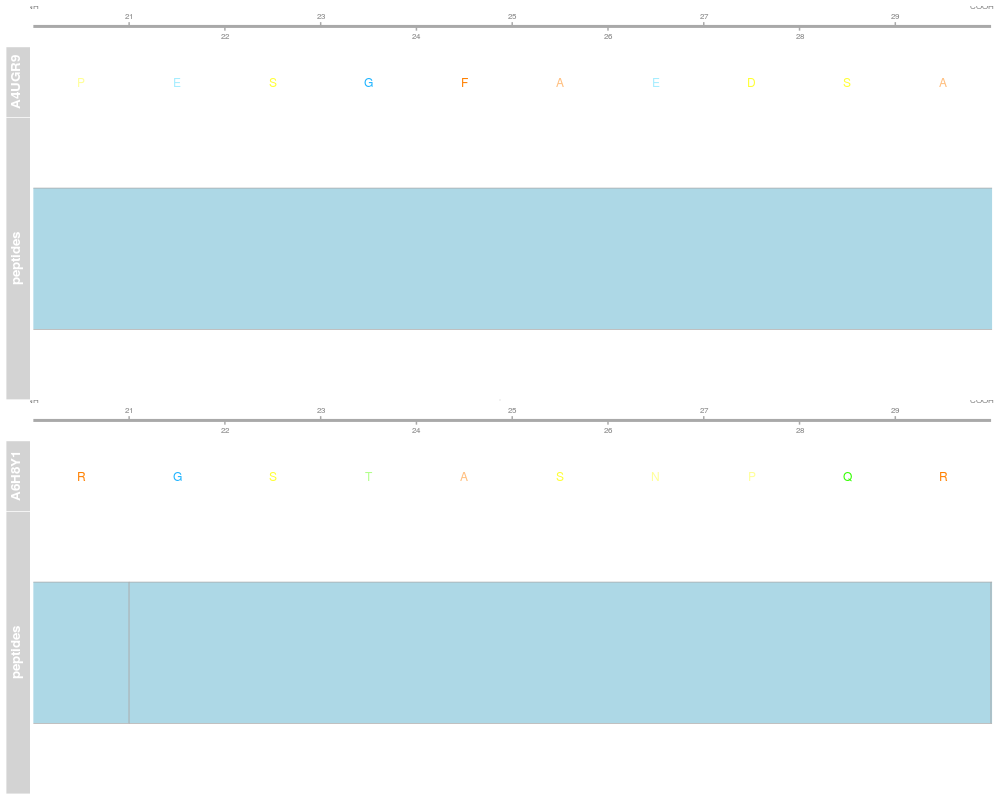
plot(p[1])
plot(p[1], # the first protein has 36 peptides
fill = c(rep("orange", 13), rep("steelblue", 13)))
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(Pbase)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Rcpp
Loading required package: Gviz
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: grid
This is Pbase version 0.12.2
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/Pbase/Proteins-class.Rd_%03d_medium.png", width=480, height=480)
> ### Name: Proteins-class
> ### Title: The 'Proteins' Class for Proteomics Data And Meta-Data
> ### Aliases: Proteins-class class:Proteins Proteins
> ### Proteins,character,missing-method Proteins,missing,character-method
> ### Proteins,missing,missing-method [,Proteins,ANY,ANY-method
> ### [,Proteins,ANY,ANY,ANY-method [[,Proteins,ANY,ANY-method
> ### aa,Proteins-method aa ametadata,Proteins-method ametadata acols
> ### metadata,Proteins-method metadata<-,Proteins-method
> ### pfeatures,Proteins-method pfeatures plot,Proteins,missing-method
> ### pmetadata,Proteins-method pmetadata pcols pranges,Proteins-method
> ### pranges pranges<-,Proteins,CompressedIRangesList-method pranges<-
> ### pfilter,Proteins-method pfilter pvarLabels,Proteins-method pvarLabels
> ### avarLabels,Proteins-method avarLabels length,Proteins-method
> ### seqnames,Proteins-method show,Proteins-method cleave,Proteins-method
> ### addIdentificationData,Proteins,character-method addPeptideFragments
> ### addPeptideFragments,Proteins,character-method rmEmptyRanges
> ### proteotypic proteinCoverage isCleaved
> ### Keywords: classes
>
> ### ** Examples
>
>
> ## Create a Protein object reading all proteins from a fasta file.
> fastaFiles <- list.files(system.file("extdata", package = "Pbase"),
+ pattern = "fasta", full.names = TRUE)
> p <- Proteins(fastaFiles)
> p
S4 class type : Proteins
Class version : 0.1
Created : Wed Jul 6 02:39:44 2016
Number of Proteins: 963
Sequences:
[1] A4UGR9 [2] A6H8Y1 ... [962] Pb98 [963] Pb99
Sequence features:
[1] DB [2] AccessionNumber ... [10] Comment [11] Filename
Peptide features: None
> metadata(p)
$created
[1] "Wed Jul 6 02:39:44 2016"
>
> ## Adding custom metadata
> metadata(p, "Comment") <- "I love R"
> metadata(p)
$created
[1] "Wed Jul 6 02:39:44 2016"
$Comment
[1] "I love R"
>
> ## Plotting
> plot(p[1:5], from = 1, to = 30)
>
> ## Cleaving
> pp <- cleave(p[1:100])
> pp <- proteotypic(pp)
> pp
S4 class type : Proteins
Class version : 0.1
Created : Wed Jul 6 02:39:44 2016
Number of Proteins: 100
Sequences:
[1] A4UGR9 [2] A6H8Y1 ... [99] B1X737 [100] B1X740
Sequence features:
[1] DB [2] AccessionNumber ... [10] Comment [11] Filename
Peptide features:
MissedCleavages, Proteotypic
> pcols(pp[1:2])
SplitDataFrameList of length 2
$A4UGR9
DataFrame with 457 rows and 2 columns
MissedCleavages Proteotypic
<Rle> <Rle>
1 0 TRUE
2 0 TRUE
3 0 TRUE
4 0 FALSE
5 0 FALSE
... ... ...
453 0 TRUE
454 0 TRUE
455 0 TRUE
456 0 TRUE
457 0 TRUE
...
<1 more element>
>
> plot(pp[1:2], from = 20, to = 30)
>
> ## Protein coverage
> pp <- proteinCoverage(pp)
> avarLabels(pp)
[1] "DB" "AccessionNumber" "EntryName" "IsoformName"
[5] "ProteinName" "OrganismName" "GeneName" "ProteinExistence"
[9] "SequenceVersion" "Comment" "Filename" "Coverage"
> acols(pp)$Coverage
A4UGR9 A6H8Y1 B1X5X3 B1X5X8 B1X5Y6 B1X5Y8 B1X603 B1X604 B1X620 B1X626 B1X637
1 1 1 1 1 1 1 1 1 1 1
B1X638 B1X641 B1X645 B1X650 B1X655 B1X662 B1X666 B1X669 B1X677 B1X678 B1X6A7
1 1 1 1 1 1 1 1 1 1 1
B1X6A8 B1X6B7 B1X6B9 B1X6C7 B1X6D9 B1X6E0 B1X6E6 B1X6E7 B1X6E8 B1X6E9 B1X6F0
1 1 1 1 1 1 1 1 1 1 1
B1X6F3 B1X6F4 B1X6F5 B1X6F6 B1X6F7 B1X6F8 B1X6F9 B1X6G0 B1X6G1 B1X6G2 B1X6G3
1 1 1 1 1 1 1 1 1 1 1
B1X6G4 B1X6G5 B1X6G6 B1X6G7 B1X6G8 B1X6G9 B1X6H0 B1X6H1 B1X6H2 B1X6H7 B1X6I9
1 1 1 1 1 1 1 1 1 1 1
B1X6J0 B1X6J1 B1X6J5 B1X6J6 B1X6K7 B1X6L0 B1X6L1 B1X6L3 B1X6L4 B1X6L6 B1X6L7
1 1 1 1 1 1 1 1 1 1 1
B1X6M1 B1X6M8 B1X6P1 B1X6Q1 B1X6Q4 B1X6Q5 B1X6Q6 B1X6Q7 B1X6Q8 B1X6Q9 B1X6S1
1 1 1 1 1 1 1 1 1 1 1
B1X6S3 B1X6S5 B1X6U3 B1X6V8 B1X6V9 B1X6W2 B1X6W3 B1X6W6 B1X6X2 B1X6X3 B1X6X5
1 1 1 1 1 1 1 1 1 1 1
B1X6X7 B1X6Y8 B1X6Z8 B1X703 B1X705 B1X709 B1X711 B1X720 B1X731 B1X733 B1X737
1 1 1 1 1 1 1 1 1 1 1
B1X740
1
> pp
S4 class type : Proteins
Class version : 0.1
Created : Wed Jul 6 02:39:44 2016
Number of Proteins: 100
Sequences:
[1] A4UGR9 [2] A6H8Y1 ... [99] B1X737 [100] B1X740
Sequence features:
[1] DB [2] AccessionNumber ... [11] Filename [12] Coverage
Peptide features:
MissedCleavages, Proteotypic
>
>
> ## Add indentification data
> idfile <- system.file("extdata/Thermo_Hela_PRTC_selected.mzid",
+ package = "Pbase")
> p <- addIdentificationData(p, idfile)
Reading 1 identification files:
1. /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
done.
> pranges(p)
IRangesList of length 9
$A4UGR9
IRanges object with 36 ranges and 28 metadata columns:
start end width | DB AccessionNumber EntryName
<integer> <integer> <integer> | <Rle> <character> <character>
A4UGR9 2743 2760 18 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 307 318 12 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 1858 1870 13 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 1699 1708 10 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 2622 2637 16 | sp A4UGR9 XIRP2_HUMAN
... ... ... ... . ... ... ...
A4UGR9 20 31 12 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 1712 1729 18 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 48 61 14 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 2082 2094 13 | sp A4UGR9 XIRP2_HUMAN
A4UGR9 2743 2756 14 | sp A4UGR9 XIRP2_HUMAN
IsoformName
<Rle>
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
... ...
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
A4UGR9 <NA>
ProteinName
<character>
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
... ...
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
A4UGR9 sp|A4UGR9|XIRP2_HUMAN Xin actin-binding repeat-containing protein 2
OrganismName GeneName ProteinExistence SequenceVersion
<Rle> <Rle> <Rle> <Rle>
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
... ... ... ... ...
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
A4UGR9 Homo sapiens XIRP2 Evidence at protein level 2
Comment spectrumID chargeState rank passThreshold
<Rle> <factor> <integer> <integer> <logical>
A4UGR9 <NA> index=124 3 1 TRUE
A4UGR9 <NA> index=28 2 1 TRUE
A4UGR9 <NA> index=20 2 1 TRUE
A4UGR9 <NA> index=187 2 1 TRUE
A4UGR9 <NA> index=211 3 1 TRUE
... ... ... ... ... ...
A4UGR9 <NA> index=99 2 1 TRUE
A4UGR9 <NA> index=9 2 1 TRUE
A4UGR9 <NA> index=122 2 1 TRUE
A4UGR9 <NA> index=87 2 1 TRUE
A4UGR9 <NA> index=77 2 1 TRUE
experimentalMassToCharge calculatedMassToCharge sequence
<numeric> <numeric> <factor>
A4UGR9 715.0305 715.0308 QEITQNKSFFSSVKESQR
A4UGR9 715.9177 715.4117 LPVPKDVYSKQR
A4UGR9 786.9066 786.9081 EQNNDALEKSLRR
A4UGR9 629.8380 629.3386 SLKESSHRWK
A4UGR9 645.3429 645.3511 LKMVPRKQREFSGSDR
... ... ... ...
A4UGR9 619.2888 618.7782 PESGFAEDSAAR
A4UGR9 1014.0198 1013.5117 QPDAIPGDIEKAIECLEK
A4UGR9 821.4005 820.8909 MARYQAAVSRGDCR
A4UGR9 720.3445 720.3527 TNTSTGLKMAMER
A4UGR9 821.9231 821.9254 QEITQNKSFFSSVK
modNum isDecoy post pre start end
<integer> <logical> <factor> <factor> <integer> <integer>
A4UGR9 0 FALSE D K 2743 2760
A4UGR9 0 FALSE N R 307 318
A4UGR9 0 FALSE L R 1858 1870
A4UGR9 0 FALSE E K 1699 1708
A4UGR9 0 FALSE G K 2622 2637
... ... ... ... ... ... ...
A4UGR9 0 FALSE G K 20 31
A4UGR9 1 FALSE A K 1712 1729
A4UGR9 1 FALSE S R 48 61
A4UGR9 0 FALSE S K 2082 2094
A4UGR9 0 FALSE E K 2743 2756
DatabaseAccess DBseqLength DatabaseSeq acquisitionNum
<factor> <integer> <factor> <numeric>
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 124
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 28
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 20
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 187
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 211
... ... ... ... ...
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 99
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 9
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 122
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 87
A4UGR9 sp|A4UGR9|XIRP2_HUMAN 3374 77
filenames
<Rle>
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
... ...
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
A4UGR9 /home/ddbj/local/lib64/R/library/Pbase/extdata/Thermo_Hela_PRTC_selected.mzid
...
<8 more elements>
> pfeatures(p)
AAStringSetList of length 9
[["A4UGR9"]] A4UGR9=QEITQNKSFFSSVKESQR ... A4UGR9=QEITQNKSFFSSVK
[["A6H8Y1"]] A6H8Y1=EDAEQVALEVDLNQKKRR ... A6H8Y1=ARLSVKPNVRPGVGARGSTASNPQRGR
[["O43707"]] O43707=QQRKTFTAWCNSHLR ... O43707=VGWEQLLTTIAR
[["O75369"]] O75369=DLDIIDNYDYSHTVK ... O75369=VQAQGPGLKEAFTNK
[["P00558"]] P00558=ELNYFAKALESPER P00558=DLMSKAEK ... P00558=GTKALMDEVVK
[["P02545"]] P02545=METPSQRRATR ... P02545=RATRSGAQASSTPLSPTR
[["P04075"]] P04075=GVVPLAGTNGETTTQGLDGLSER ... P04075=YTPSGQAGAAASESLFVSNHAY
[["P04075-2"]] P04075-2=GVVPLAGTNGETTTQGLDGLSER ...
[["P60709"]] P60709=DLTDYLMKILTER
>
> plot(p[1])
> plot(p[1], # the first protein has 36 peptides
+ fill = c(rep("orange", 13), rep("steelblue", 13)))
>
>
>
>
>
> dev.off()
null device
1
>
 .
.