Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
The Polyfit extension to the DESeq functions nbinomTest() and nbinomTestForMatrices()DescriptionPolyfit extensions to the DESeq functions UsagepfNbinomTest(cds, condA, condB, pvals_only = FALSE, eps = NULL) pfNbinomTestForMatrices(countsA, countsB, sizeFactorsA, sizeFactorsB, dispsA, dispsB ) Arguments
DetailsThese functions have the same behaviour as the DESeq functions Value
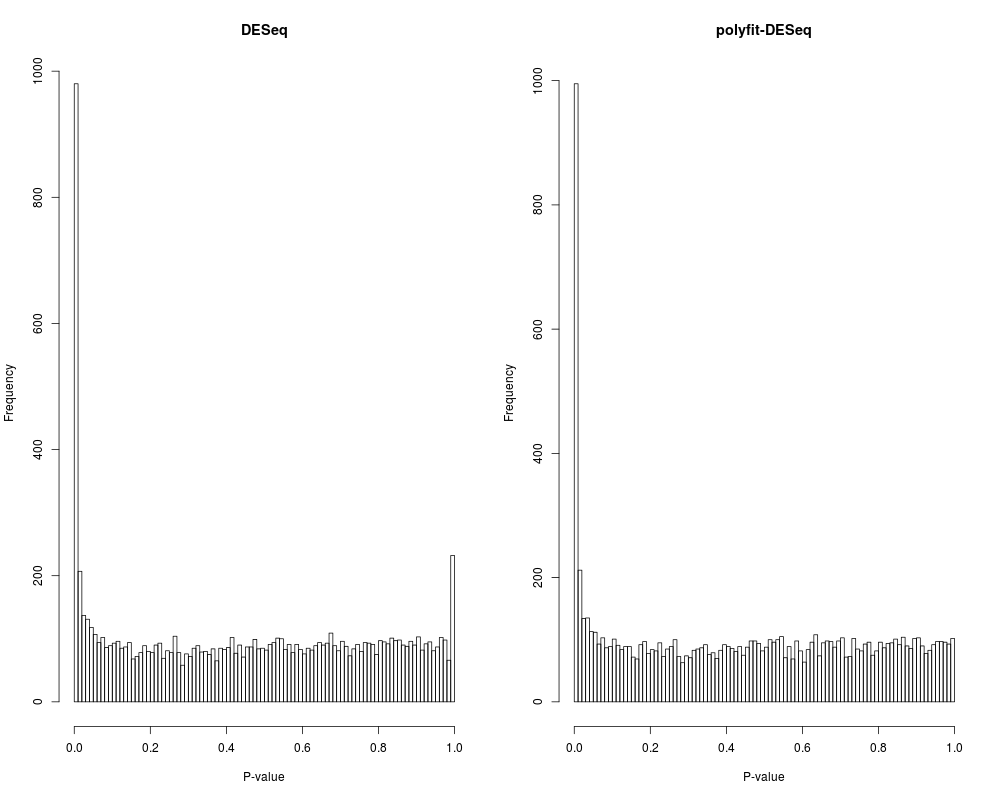
Author(s)Conrad Burden, conrad.burden@anu.edu.au, based on software by Simon Anders ReferencesBurden, C.J., Qureshi, S. and Wilson, S.R. (2014). Error estimates for the analysis of differential expression from RNA-seq count data, PeerJ PrePrints 2:e400v1. Anders, S. and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biology, 11(10), R106. Examplescds <- makeExampleCountDataSet() cds <- estimateSizeFactors( cds ) cds <- estimateDispersions( cds ) nbT <- nbinomTest( cds, "A", "B" ) head( nbT ) nbTPolyfit <- pfNbinomTest( cds, "A", "B" ) head( nbTPolyfit ) oldpar <- par(mfrow=c(1,2)) hist(nbT$pval,breaks=seq(0,1,by=0.01), xlab="P-value", main="DESeq") hist(nbTPolyfit$pval,breaks=seq(0,1,by=0.01), xlab="P-value", main="polyfit-DESeq") par(oldpar) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(Polyfit)
Loading required package: DESeq
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: locfit
locfit 1.5-9.1 2013-03-22
Loading required package: lattice
Welcome to 'DESeq'. For improved performance, usability and
functionality, please consider migrating to 'DESeq2'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/Polyfit/pfNbinomTest.Rd_%03d_medium.png", width=480, height=480)
> ### Name: pfNbinomTest
> ### Title: The Polyfit extension to the DESeq functions nbinomTest() and
> ### nbinomTestForMatrices()
> ### Aliases: pfNbinomTest pfNbinomTestForMatrices
>
> ### ** Examples
>
> cds <- makeExampleCountDataSet()
> cds <- estimateSizeFactors( cds )
> cds <- estimateDispersions( cds )
> nbT <- nbinomTest( cds, "A", "B" )
> head( nbT )
id baseMean baseMeanA baseMeanB foldChange log2FoldChange pval
1 gene_1_T 691.17043 316.71443 940.80777 2.9705239 1.57071738 0.1181887
2 gene_2_F 19.28415 19.68099 19.01959 0.9663942 -0.04931635 0.8356644
3 gene_3_F 61.69449 73.43875 53.86498 0.7334680 -0.44719406 0.5890611
4 gene_4_F 260.09763 232.04474 278.79956 1.2014905 0.26482526 0.7905044
5 gene_5_F 692.91586 1046.12178 457.44526 0.4372773 -1.19337980 0.1730757
6 gene_6_F 164.81349 206.63188 136.93456 0.6626982 -0.59357620 0.4102879
padj
1 0.5717126
2 1.0000000
3 0.9735589
4 1.0000000
5 0.6889195
6 0.9004707
> nbTPolyfit <- pfNbinomTest( cds, "A", "B" )
> head( nbTPolyfit )
id baseMean baseMeanA baseMeanB foldChange log2FoldChange pval
1 gene_1_T 691.17043 316.71443 940.80777 2.9705239 1.57071738 0.1180088
2 gene_2_F 19.28415 19.68099 19.01959 0.9663942 -0.04931635 0.8140754
3 gene_3_F 61.69449 73.43875 53.86498 0.7334680 -0.44719406 0.5789490
4 gene_4_F 260.09763 232.04474 278.79956 1.2014905 0.26482526 0.7882567
5 gene_5_F 692.91586 1046.12178 457.44526 0.4372773 -1.19337980 0.1724955
6 gene_6_F 164.81349 206.63188 136.93456 0.6626982 -0.59357620 0.4068913
padj
1 0.5645545
2 0.9952024
3 0.9576691
4 0.9909575
5 0.6783692
6 0.8843828
>
> oldpar <- par(mfrow=c(1,2))
> hist(nbT$pval,breaks=seq(0,1,by=0.01),
+ xlab="P-value", main="DESeq")
> hist(nbTPolyfit$pval,breaks=seq(0,1,by=0.01),
+ xlab="P-value", main="polyfit-DESeq")
> par(oldpar)
>
>
>
>
>
> dev.off()
null device
1
>
|