This function takes as input tumor and normal control coverage and
allelic fractions of germline variants and somatic mutations.
Coverage data is provided in GATK DepthOfCoverage format, allelic fraction
in VCF format (e.g. obtained by MuTect). Normal control does not need to
be matched (from the same patient). In case VCF does not contain somatic
status, it should contain dbSNP and optionally COSMIC annotation.
Returns purity and ploidy combinations, sorted by likelihood score.
Provides copy number and LOH data, by both gene and genomic region.
GATK coverage file of normal control (optional if
log.ratio is provided - then it will be only used to filter low coverage
exons). Should be already GC-normalized. Needs to be either a file name
or data read with the readCoverageGatk function.
gatk.tumor.file
GATK coverage file of tumor. Should be already
GC-normalized. Needs to be either a file name or data read with the
readCoverageGatk function.
log.ratio
Copy number log-ratios for all exons in the coverage files.
If NULL, calculated based on coverage files.
seg.file
Segmented data. Optional, to support matched SNP6 data.
If null, use coverage files or log.ratio to segment the data.
seg.file.sdev
If seg.file provided, the log-ratio standard deviation,
used to model likelihood of sub-clonal copy number events.
vcf.file
VCF file, tested with MuTect output files. Optional, but
typically needed to select between local optima of similar likelihood. Can
also be a CollapsedVCF, read with the readVcf function. Requires a DB info
flag for dbSNP membership. The default fun.setPriorVcf function will also
look for a Cosmic.CNT slot, containing the hits in the COSMIC database.
Again, do not expect very useful results without a VCF file.
genome
Genome version, required for the readVcf function.
sex
Sex of sample. If ?, detect.
fun.filterVcf
Function for filtering variants. Expected output is a
list with elements vcf (CollapsedVCF), flag (TRUE/FALSE) and flag_comment
(string). The flags will be added to the output data and can be used to
warn users, for example when samples look too noisy. Default filter will
remove variants flagged by MuTect, but will keep germline variants. If
ran in matched normal mode, it will by default use somatic status of
variants and filter non-somatic calls with allelic fraction significantly
different from 0.5 in normal.
args.filterVcf
Arguments for variant filtering function. Arguments
vcf, tumor.id.in.vcf, coverage.cutoff and verbose are required in the
filter function and are automatically set (do NOT set them here again).
fun.setPriorVcf
Function to set prior for somatic status for each
variant in the VCF.
args.setPriorVcf
Arguments for somatic prior function.
fun.segmentation
Function for segmenting the copy number log-ratios.
Expected return value is a list with elements seg (the segmentation) and
size (the size in bp for all segments).
args.segmentation
Arguments for segmentation function. Arguments
normal, tumor, log.ratio, plot.cnv, coverage.cutoff, sampleid, vcf,
tumor.id.in.vcf, verbose are required in the segmentation function and
automatically set (do NOT set them here again).
fun.focal
Function for identifying focal amplifications.
args.focal
Arguments for focal amplification function.
sampleid
Sample id, provided in output files etc.
min.ploidy
Minimum ploidy to be considered.
max.ploidy
Maximum ploidy to be considered.
test.num.copy
Copy numbers tested in the grid search. Note that focal
amplifications can have much higher copy numbers, but they will be labeled
as subclonal (because they do not fit the integer copy numbers).
test.purity
Considered tumor purity values.
prior.purity
Priors for purity if they are available. Only change
when you know what you are doing.
max.candidate.solutions
Number of local optima considered in optimization
and variant fitting steps. If there are too many local optima, it will use
specified number of top candidate solutions, but will also include all
optima close to diploid, because silent genomes have often lots of local
optima.
candidates
Candidates to optimize from a previous run
(return.object$candidates).
If NULL, do 2D grid search and find local optima.
coverage.cutoff
Minimum exon coverage in both normal and tumor. Exons
with lower coverage are ingored. The cutoff choice depends on the expected
purity and overall coverage. High purity samples might need a lower cutoff
to call homozygous deletions. If an exon.weigh.file (below) is NOT
specified, it is recommended to set a higher cutoff (e.g. 20) to remove
noise from unreliable exon measurements.
max.non.clonal
Maximum genomic fraction assigned to a subclonal copy
number state.
max.homozygous.loss
Maximum genomic fraction assigned to homozygous loss.
This is set to a fairly high default value to not exclude correct
solutions, especially in noisy segmentations.
iterations
Maximum number of iterations in the Simulated Annealing copy
number fit optimization.
log.ratio.calibration
re-calibrate log-ratios in the window
sd(log.ratio)*log.ratio.calibration.
gc.gene.file
A mapping file that assigns GC content and gene symbols
to each exon in the coverage files. Used for generating gene level calls.
First column in format CHR:START-END. Second column GC content (0 to 1).
Third column gene symbol.
filter.lowhigh.gc.exons
Quantile q (defines lower q and upper 1-q)
for removing exons with outlier GC profile. Assuming that GC correction
might not have been worked on those. Requires gc.gene.file.
filter.targeted.base
Exclude exons with targeted base (size) smaller
than this cutoff. This is useful when the same interval file was used to
calculate GC content. For such small exons, the GC content is likely
very different from the true GC content of the probes.
max.logr.sdev
Flag noisy samples with segment log-ratio standard deviation
larger than this. Assay specific and needs to be calibrated.
max.segments
Flag noisy samples with a large number of segments. Assay
specific and needs to be calibrated.
plot.cnv
Generate segmentation plots.
verbose
Verbose output.
post.optimize
Optimize purity using final SCNA-fit and SNVs. This
might take a long time when lots of SNVs need to be fitted, but will
typically result in a slightly more accurate purity, especially for rather
silent genomes or very low purities. Otherwise, it will just use the
purity determined via the SCNA-fit.
...
Additional parameters passed to the segmentation function.
Value
A list with elements
candidates
Results of the grid search.
results
All local optima, sorted by final rank.
input
The input data.
Author(s)
Markus Riester
Examples
gatk.normal.file <- system.file("extdata", "example_normal.txt",
package="PureCN")
gatk.tumor.file <- system.file("extdata", "example_tumor.txt",
package="PureCN")
vcf.file <- system.file("extdata", "example_vcf.vcf",
package="PureCN")
gc.gene.file <- system.file("extdata", "example_gc.gene.file.txt",
package="PureCN")
# Speed-up the runAbsoluteCN call by using the stored grid-search
# (purecn.example.output$candidates).
data(purecn.example.output)
# The max.candidate.solutions parameter is set to a very low value only to
# speed-up this example. This is not a good idea for real samples.
ret <-runAbsoluteCN(gatk.normal.file=gatk.normal.file,
gatk.tumor.file=gatk.tumor.file,
candidates=purecn.example.output$candidates, max.candidate.solutions=2,
vcf.file=vcf.file, sampleid='Sample1', gc.gene.file=gc.gene.file)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(PureCN)
Loading required package: DNAcopy
Loading required package: VariantAnnotation
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: GenomeInfoDb
Loading required package: stats4
Loading required package: S4Vectors
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: Rsamtools
Loading required package: Biostrings
Loading required package: XVector
Attaching package: 'VariantAnnotation'
The following object is masked from 'package:base':
tabulate
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/PureCN/runAbsoluteCN.Rd_%03d_medium.png", width=480, height=480)
> ### Name: runAbsoluteCN
> ### Title: Run PureCN implementation of ABSOLUTE
> ### Aliases: runAbsoluteCN
>
> ### ** Examples
>
> gatk.normal.file <- system.file("extdata", "example_normal.txt",
+ package="PureCN")
> gatk.tumor.file <- system.file("extdata", "example_tumor.txt",
+ package="PureCN")
> vcf.file <- system.file("extdata", "example_vcf.vcf",
+ package="PureCN")
> gc.gene.file <- system.file("extdata", "example_gc.gene.file.txt",
+ package="PureCN")
>
> # Speed-up the runAbsoluteCN call by using the stored grid-search
> # (purecn.example.output$candidates).
> data(purecn.example.output)
>
> # The max.candidate.solutions parameter is set to a very low value only to
> # speed-up this example. This is not a good idea for real samples.
>
> ret <-runAbsoluteCN(gatk.normal.file=gatk.normal.file,
+ gatk.tumor.file=gatk.tumor.file,
+ candidates=purecn.example.output$candidates, max.candidate.solutions=2,
+ vcf.file=vcf.file, sampleid='Sample1', gc.gene.file=gc.gene.file)
Loading GATK coverage files...
Sex of sample: ?
Removing 7 small exons.
Removing 15 low/high GC exons.
Loading VCF...
Assuming LIB-02240e4 is tumor in VCF file.
Found 2331 variants in VCF file.
Removing 0 non heterozygous (in matched normal) germline SNPs.
Removing 62 SNPs with AF < 0.03 or AF >= 0.97 or less than 3 supporting reads or depth < 15.
Found SOMATIC annotation in VCF. Setting somatic prior probabilities for somatic variants to 0.999 or to 1e-04 otherwise.
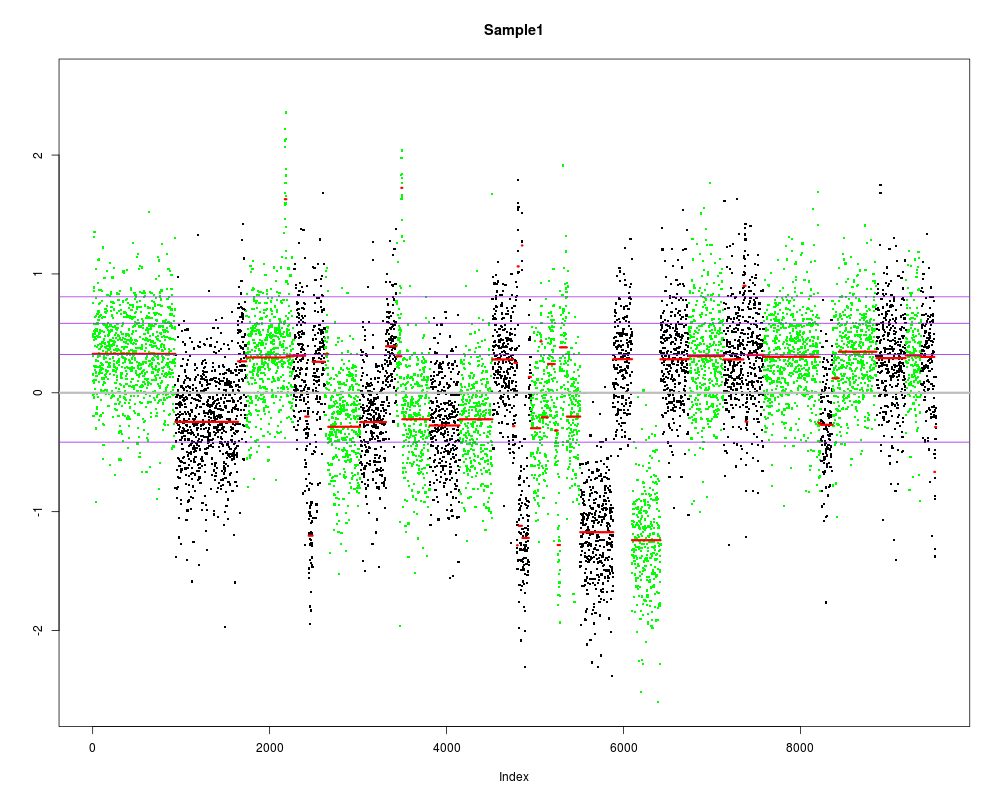
Segmenting data...
Removing 267 low coverage exons.
Analyzing: Sample1
Setting multi-figure configuration
Call:
segment(x = smoothed.CNA.obj, alpha = alpha, undo.splits = undo.splits,
undo.SD = sdundo, verbose = ifelse(verbose, 1, 0))
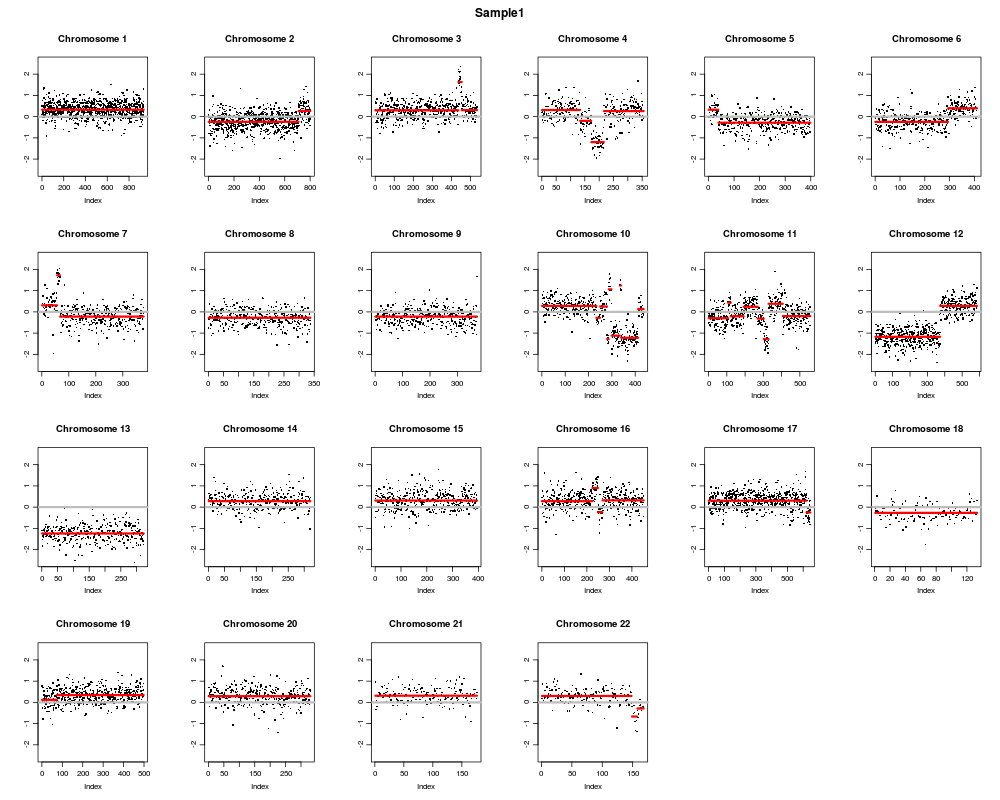
ID chrom loc.start loc.end num.mark seg.mean
1 Sample1 1 1216044.5 248722319 934 0.3272
2 Sample1 2 1638036.0 231775198 708 -0.2446
3 Sample1 2 236403412.5 241737117 93 0.2648
4 Sample1 3 11832017.5 149470198 436 0.2969
5 Sample1 3 150264604.0 151542537 18 1.6288
6 Sample1 3 151545662.5 195938114 80 0.3072
7 Sample1 4 843512.0 70146580 133 0.3118
8 Sample1 4 75673305.5 77700146 39 -0.1997
9 Sample1 4 81188156.5 108831608 44 -1.2019
10 Sample1 4 110635592.5 186611721 139 0.2606
11 Sample1 5 442758.0 10761154 38 0.3255
12 Sample1 5 38869183.5 180687408 360 -0.2858
13 Sample1 6 2623865.0 144219759 293 -0.2464
14 Sample1 6 144224235.5 170862274 117 0.3914
15 Sample1 7 938572.5 14028656 56 0.3075
16 Sample1 7 23286512.0 23313764 11 1.7239
17 Sample1 7 26232167.0 156469232 310 -0.2223
18 Sample1 8 6264200.0 145537891 337 -0.2747
19 Sample1 9 214953.0 139440208 371 -0.2226
20 Sample1 10 323391.5 72576624 233 0.2804
21 Sample1 10 72604313.0 72645621 16 -0.2807
22 Sample1 10 72648289.5 75000741 30 0.2522
23 Sample1 10 82300671.5 82403794 7 -1.2827
24 Sample1 10 85982056.5 88768888 12 1.0633
25 Sample1 10 91066426.0 99790218 36 -1.1192
26 Sample1 10 102283640.5 102289566 5 1.2394
27 Sample1 10 103541552.5 121214530 73 -1.2211
28 Sample1 10 124591880.5 134121207 24 0.1328
29 Sample1 11 2291272.0 34378690 106 -0.2981
30 Sample1 11 36614927.0 44081430 15 0.4322
31 Sample1 11 46880700.5 57317514 71 -0.2062
32 Sample1 11 57947383.5 65172438 77 0.2423
33 Sample1 11 65340286.5 66335024 33 -0.3169
34 Sample1 11 66335504.5 71209486 26 -1.2807
35 Sample1 11 71847083.0 82549523 78 0.3824
36 Sample1 11 82550385.5 134134828 151 -0.2000
37 Sample1 12 1740561.0 99126272 373 -1.1729
38 Sample1 12 113537804.0 124428836 212 0.2830
39 Sample1 13 20398996.5 114438189 322 -1.2399
40 Sample1 14 20757846.0 101349088 319 0.2821
41 Sample1 15 27216709.5 99926272 394 0.3090
42 Sample1 16 230533.5 31123514 224 0.2804
43 Sample1 16 56899289.0 56947247 25 0.9000
44 Sample1 16 57507348.5 57722319 20 -0.2426
45 Sample1 16 66918983.0 90038049 183 0.3186
46 Sample1 17 1399145.5 76832320 621 0.3029
47 Sample1 17 77768896.0 80559278 24 -0.2538
48 Sample1 18 5394737.5 71825664 133 -0.2692
49 Sample1 19 1481982.5 3730529 73 0.1224
50 Sample1 19 3731985.0 57301280 425 0.3454
51 Sample1 20 207959.0 62610776 330 0.2918
52 Sample1 21 11098731.0 47865219 176 0.3147
53 Sample1 22 17443695.0 45996257 148 0.3021
54 Sample1 22 50703417.0 50705856 9 -0.6659
55 Sample1 22 50706027.5 51066096 11 -0.2871
Mean standard deviation of log-ratios: 0.4
Optimizing purity and ploidy. Will take a minute or two...
Local optima: 0.65/1.6, 0.5/2.4, 0.9/2.4, 0.5/2
Testing local optimum at purity 0.65 and total ploidy 1.6.
Fitting SNVs for purity 0.65 and tumor ploidy 1.38.
Analyzing: Sample1
Optimized purity: 0.65
Testing local optimum at purity 0.5 and total ploidy 2.4.
Fitting SNVs for purity 0.48 and tumor ploidy 2.76.
Analyzing: Sample1
Optimized purity: 0.48
Testing local optimum at purity 0.9 and total ploidy 2.4.
Fitting SNVs for purity 0.95 and tumor ploidy 2.38.
Analyzing: Sample1
Optimized purity: 0.95
Testing local optimum at purity 0.5 and total ploidy 2.
Fitting SNVs for purity 0.48 and tumor ploidy 2.76.
Analyzing: Sample1
Optimized purity: 0.48
Remember, posterior probabilities assume a correct SCNA fit.
Warning message:
In runAbsoluteCN(gatk.normal.file = gatk.normal.file, gatk.tumor.file = gatk.tumor.file, :
Too many candidate solutions! Trying optimizing the top candidates.
>
>
>
>
>
> dev.off()
null device
1
>
 .
.