|
Last data update: 2014.03.03
|
R: An R package for RNA visualization and analysis
| R4RNA-package | R Documentation |
An R package for RNA visualization and analysis
Description
An R package for RNA visualization and analysis
Examples
# Read input data
predicted <- readHelix(system.file("extdata", "helix.txt", package = "R4RNA"))
known <- readVienna(system.file("extdata", "vienna.txt", package = "R4RNA"))
sequence <- as.character(readBStringSet(system.file("extdata", "fasta.txt", package = "R4RNA")))
plotHelix(predicted)
pval.coloured <- colourByValue(predicted, log = TRUE, get = TRUE)
plotDoubleHelix(pval.coloured, known, scale = FALSE)
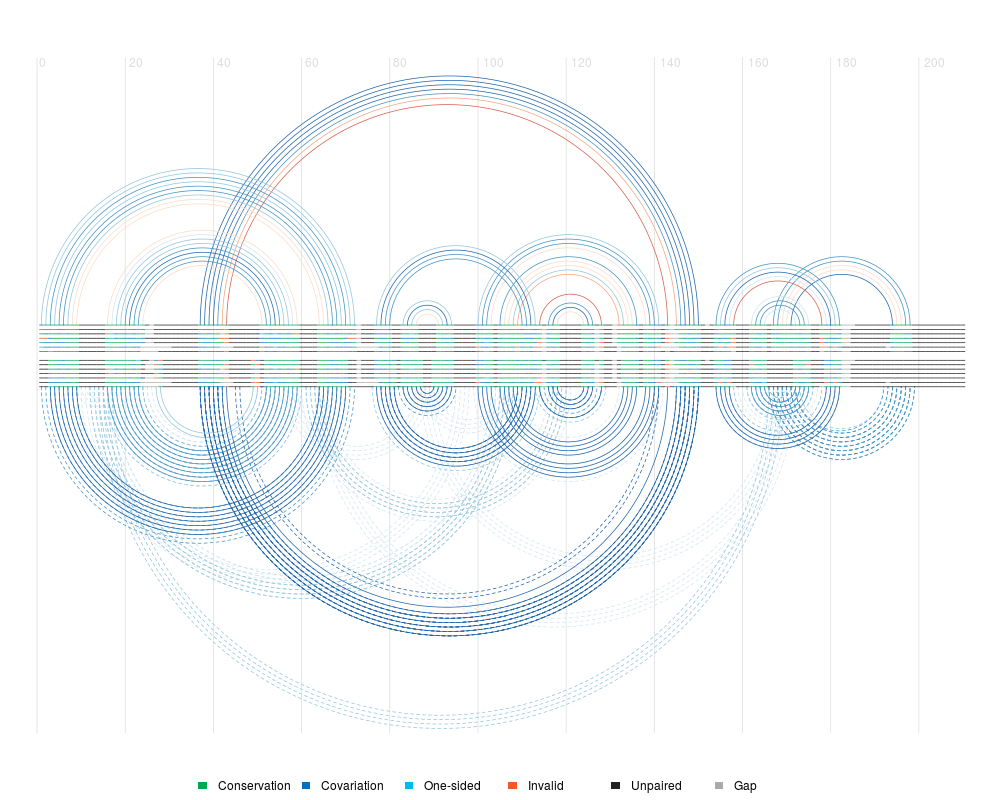
plotOverlapHelix(pval.coloured, known)
cov.coloured <- colourByCovariation(known, sequence, get = TRUE)
plotCovariance(sequence, cov.coloured)
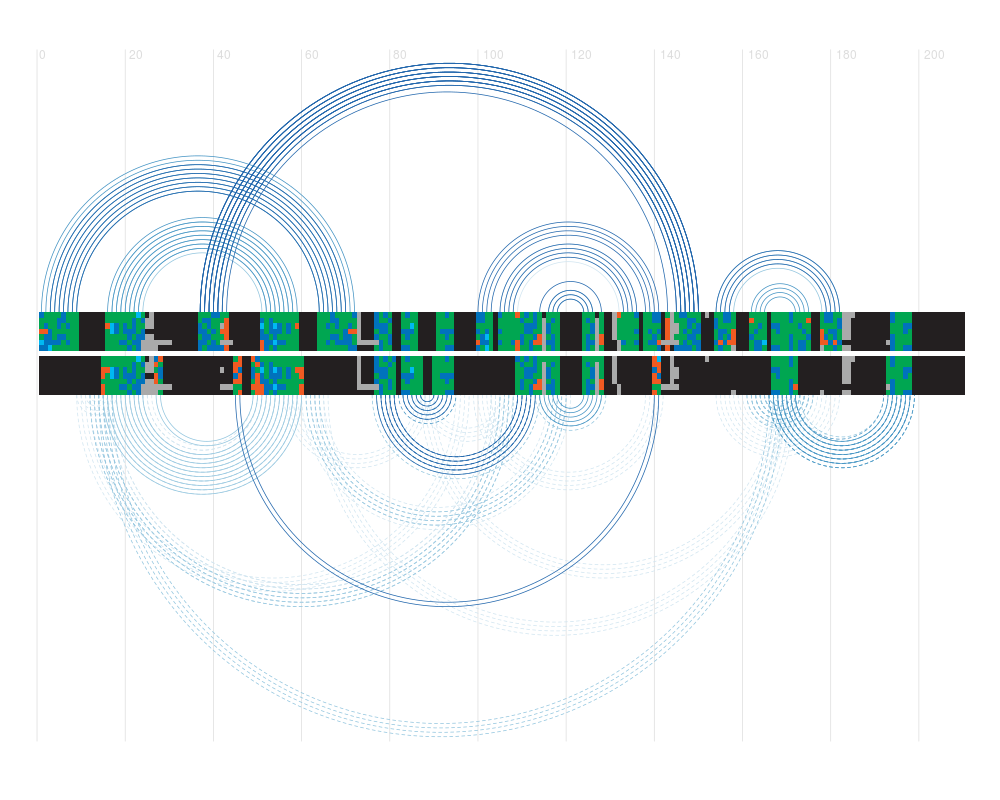
plotDoubleCovariance(cov.coloured, pval.coloured, sequence,
conflict.filter = "grey")
plotOverlapCovariance(pval.coloured, known, sequence, grid = TRUE,
conflict.filter = "grey", legend = FALSE, any = TRUE)
# List of all functions
ls("package:R4RNA")
# use example() and help() for more details on each function
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(R4RNA)
Loading required package: Biostrings
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/R4RNA/R4RNA-package.Rd_%03d_medium.png", width=480, height=480)
> ### Name: R4RNA-package
> ### Title: An R package for RNA visualization and analysis
> ### Aliases: R4RNA R4RNA-package
> ### Keywords: package
>
> ### ** Examples
>
> # Read input data
> predicted <- readHelix(system.file("extdata", "helix.txt", package = "R4RNA"))
> known <- readVienna(system.file("extdata", "vienna.txt", package = "R4RNA"))
> sequence <- as.character(readBStringSet(system.file("extdata", "fasta.txt", package = "R4RNA")))
>
> plotHelix(predicted)
> pval.coloured <- colourByValue(predicted, log = TRUE, get = TRUE)
> plotDoubleHelix(pval.coloured, known, scale = FALSE)
> plotOverlapHelix(pval.coloured, known)
>
> cov.coloured <- colourByCovariation(known, sequence, get = TRUE)
> plotCovariance(sequence, cov.coloured)
>
> plotDoubleCovariance(cov.coloured, pval.coloured, sequence,
+ conflict.filter = "grey")
> plotOverlapCovariance(pval.coloured, known, sequence, grid = TRUE,
+ conflict.filter = "grey", legend = FALSE, any = TRUE)
>
> # List of all functions
> ls("package:R4RNA")
[1] "alignmentCanonical" "alignmentConservation"
[3] "alignmentCovariation" "alignmentPercentGaps"
[5] "as.helix" "baseConservation"
[7] "basepairCanonical" "basepairConservation"
[9] "basepairCovariation" "basepairFrequency"
[11] "blankPlot" "collapseHelix"
[13] "colourByBasepairFrequency" "colourByCanonical"
[15] "colourByConservation" "colourByCount"
[17] "colourByCovariation" "colourByUnknottedGroups"
[19] "colourByValue" "defaultPalette"
[21] "expandHelix" "helixCanonical"
[23] "helixConservation" "helixCovariation"
[25] "helixToBpseq" "helixToConnect"
[27] "helixToVienna" "is.helix"
[29] "isConflictingHelix" "isDuplicatingHelix"
[31] "isOverlappingHelix" "logceiling"
[33] "logfloor" "logseq"
[35] "maxHeight" "plotArc"
[37] "plotArcs" "plotCovariance"
[39] "plotDoubleCovariance" "plotDoubleHelix"
[41] "plotHelix" "plotOverlapCovariance"
[43] "plotOverlapHelix" "readBpseq"
[45] "readConnect" "readHelix"
[47] "readVienna" "structureMismatchScore"
[49] "unknottedGroups" "viennaToHelix"
[51] "writeHelix"
> # use example() and help() for more details on each function
>
>
>
>
>
> dev.off()
null device
1
>
|
 .
.