Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Create Heatmaps for TCGA DatasetsDescriptionFunction creates heatmaps (geom_tile) for TCGA Datasets. UsageheatmapTCGA(data, x, y, fill, legend.title = "Expression", legend = "right", title = "Heatmap of expression", facet.names = NULL, tile.size = 0.1, tile.color = "white", ...) Arguments
IssuesIf you have any problems, issues or think that something is missing or is not clear please post an issue on https://github.com/RTCGA/RTCGA/issues. Note
Author(s)Marcin Kosinski, m.p.kosinski@gmail.com See AlsoRTCGA website http://rtcga.github.io/RTCGA/Visualizations.html. Other RTCGA: Examples
library(RTCGA.rnaseq)
# perfrom plot
library(dplyr)
expressionsTCGA(ACC.rnaseq, BLCA.rnaseq, BRCA.rnaseq, OV.rnaseq,
extract.cols = c("MET|4233", "ZNF500|26048", "ZNF501|115560")) %>%
rename(cohort = dataset,
MET = `MET|4233`) %>%
#cancer samples
filter(substr(bcr_patient_barcode, 14, 15) == "01") %>%
mutate(MET = cut(MET,
round(quantile(MET, probs = seq(0,1,0.25)), -2),
include.lowest = TRUE,
dig.lab = 5)) -> ACC_BLCA_BRCA_OV.rnaseq
ACC_BLCA_BRCA_OV.rnaseq %>%
select(-bcr_patient_barcode) %>%
group_by(cohort, MET) %>%
summarise_each(funs(median)) %>%
mutate(ZNF500 = round(`ZNF500|26048`),
ZNF501 = round(`ZNF501|115560`)) -> ACC_BLCA_BRCA_OV.rnaseq.medians
heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq.medians,
"cohort", "MET", "ZNF500", title = "Heatmap of ZNF500 expression")
## facet example
library(RTCGA.mutations)
library(dplyr)
mutationsTCGA(BRCA.mutations, OV.mutations, ACC.mutations, BLCA.mutations) %>%
filter(Hugo_Symbol == 'TP53') %>%
filter(substr(bcr_patient_barcode, 14, 15) == "01") %>% # cancer tissue
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) -> ACC_BLCA_BRCA_OV.mutations
mutationsTCGA(BRCA.mutations, OV.mutations, ACC.mutations, BLCA.mutations) -> ACC_BLCA_BRCA_OV.mutations_all
ACC_BLCA_BRCA_OV.rnaseq %>%
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 15)) %>%
filter(bcr_patient_barcode %in%
substr(ACC_BLCA_BRCA_OV.mutations_all$bcr_patient_barcode, 1, 15)) %>%
# took patients for which we had any mutation information
# so avoided patients without any information about mutations
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>%
# strin_length(ACC_BLCA_BRCA_OV.mutations$bcr_patient_barcode) == 12
left_join(ACC_BLCA_BRCA_OV.mutations,
by = "bcr_patient_barcode") %>% #joined only with tumor patients
mutate(TP53 = ifelse(!is.na(Variant_Classification), "Mut", "WILD")) %>%
select(-bcr_patient_barcode, -Variant_Classification, -dataset, -Hugo_Symbol) %>%
group_by(cohort, MET, TP53) %>%
summarise_each(funs(median)) %>%
mutate(ZNF501 = round(`ZNF501|115560`)) -> ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians
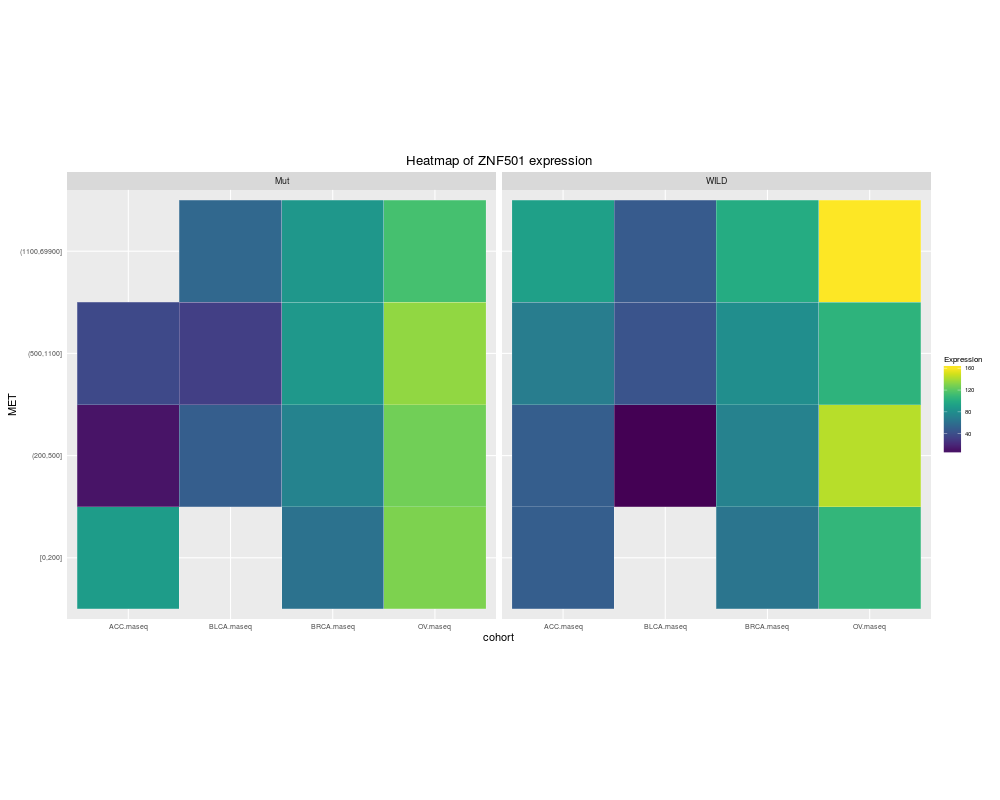
heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "cohort", "MET",
fill = "ZNF501", facet.names = "TP53", title = "Heatmap of ZNF501 expression")
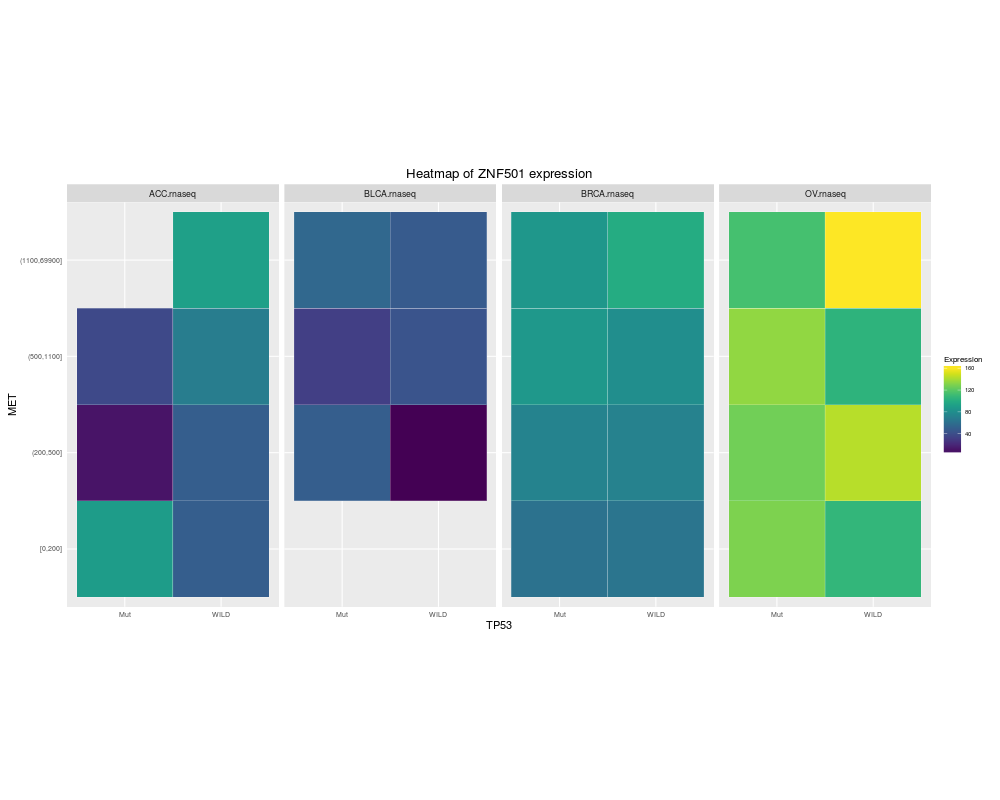
heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "TP53", "MET",
fill = "ZNF501", facet.names = "cohort", title = "Heatmap of ZNF501 expression")
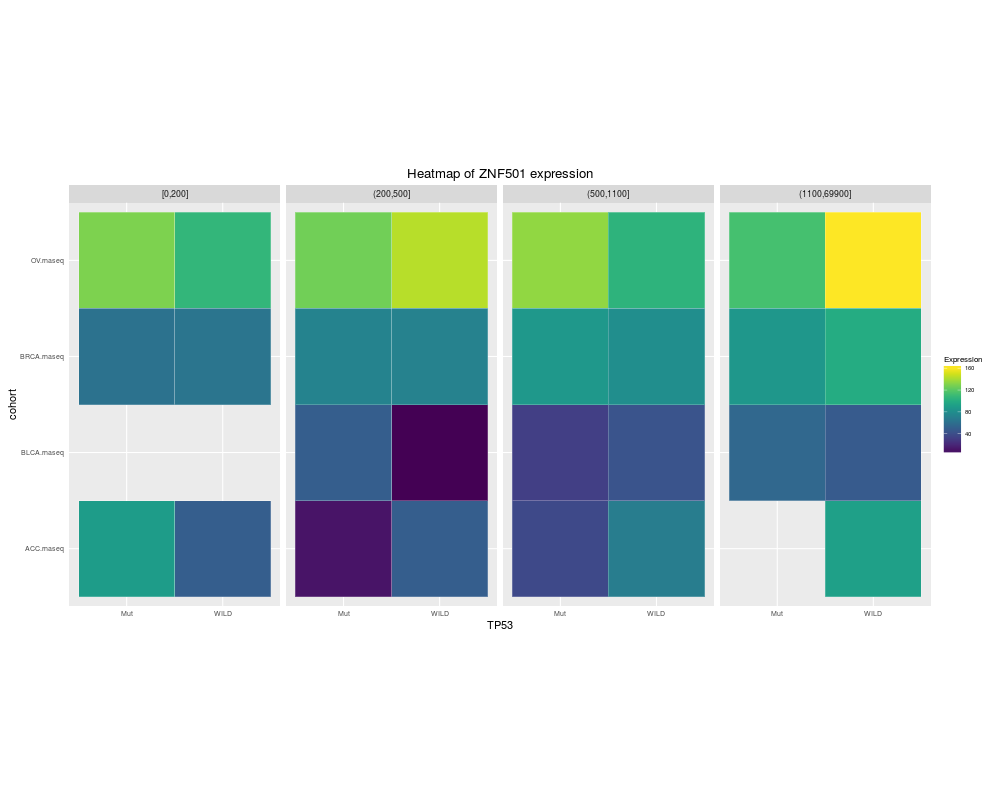
heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "TP53", "cohort",
fill = "ZNF501", facet.names = "MET", title = "Heatmap of ZNF501 expression")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(RTCGA)
Welcome to the RTCGA (version: 1.2.2).
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/RTCGA/heatmapTCGA.Rd_%03d_medium.png", width=480, height=480)
> ### Name: heatmapTCGA
> ### Title: Create Heatmaps for TCGA Datasets
> ### Aliases: heatmapTCGA
>
> ### ** Examples
>
>
>
> library(RTCGA.rnaseq)
> # perfrom plot
> library(dplyr)
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
>
>
> expressionsTCGA(ACC.rnaseq, BLCA.rnaseq, BRCA.rnaseq, OV.rnaseq,
+ extract.cols = c("MET|4233", "ZNF500|26048", "ZNF501|115560")) %>%
+ rename(cohort = dataset,
+ MET = `MET|4233`) %>%
+ #cancer samples
+ filter(substr(bcr_patient_barcode, 14, 15) == "01") %>%
+ mutate(MET = cut(MET,
+ round(quantile(MET, probs = seq(0,1,0.25)), -2),
+ include.lowest = TRUE,
+ dig.lab = 5)) -> ACC_BLCA_BRCA_OV.rnaseq
>
> ACC_BLCA_BRCA_OV.rnaseq %>%
+ select(-bcr_patient_barcode) %>%
+ group_by(cohort, MET) %>%
+ summarise_each(funs(median)) %>%
+ mutate(ZNF500 = round(`ZNF500|26048`),
+ ZNF501 = round(`ZNF501|115560`)) -> ACC_BLCA_BRCA_OV.rnaseq.medians
> heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq.medians,
+ "cohort", "MET", "ZNF500", title = "Heatmap of ZNF500 expression")
>
> ## facet example
> library(RTCGA.mutations)
> library(dplyr)
> mutationsTCGA(BRCA.mutations, OV.mutations, ACC.mutations, BLCA.mutations) %>%
+ filter(Hugo_Symbol == 'TP53') %>%
+ filter(substr(bcr_patient_barcode, 14, 15) == "01") %>% # cancer tissue
+ mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) -> ACC_BLCA_BRCA_OV.mutations
>
> mutationsTCGA(BRCA.mutations, OV.mutations, ACC.mutations, BLCA.mutations) -> ACC_BLCA_BRCA_OV.mutations_all
>
> ACC_BLCA_BRCA_OV.rnaseq %>%
+ mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 15)) %>%
+ filter(bcr_patient_barcode %in%
+ substr(ACC_BLCA_BRCA_OV.mutations_all$bcr_patient_barcode, 1, 15)) %>%
+ # took patients for which we had any mutation information
+ # so avoided patients without any information about mutations
+ mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>%
+ # strin_length(ACC_BLCA_BRCA_OV.mutations$bcr_patient_barcode) == 12
+ left_join(ACC_BLCA_BRCA_OV.mutations,
+ by = "bcr_patient_barcode") %>% #joined only with tumor patients
+ mutate(TP53 = ifelse(!is.na(Variant_Classification), "Mut", "WILD")) %>%
+ select(-bcr_patient_barcode, -Variant_Classification, -dataset, -Hugo_Symbol) %>%
+ group_by(cohort, MET, TP53) %>%
+ summarise_each(funs(median)) %>%
+ mutate(ZNF501 = round(`ZNF501|115560`)) -> ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians
>
> heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "cohort", "MET",
+ fill = "ZNF501", facet.names = "TP53", title = "Heatmap of ZNF501 expression")
> heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "TP53", "MET",
+ fill = "ZNF501", facet.names = "cohort", title = "Heatmap of ZNF501 expression")
> heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq_TP53mutations_ZNF501medians, "TP53", "cohort",
+ fill = "ZNF501", facet.names = "MET", title = "Heatmap of ZNF501 expression")
>
>
>
>
>
> dev.off()
null device
1
>
|