R: Computes a distance between two partitions of the same data
clScore
R Documentation
Computes a distance between two partitions of the same data
Description
The function takes as input two partitions of a
dataset into clusters, and returns a number which is small if the
two partitions are close, large otherwise.
Usage
clScore(c1, c2)
Arguments
c1
A vector giving the assignment of the samples to cluster for the first partition
c2
A vector giving the assignment of the samples to cluster for the second partition
Value
A number corresponding to the distance between c1 and c2
Examples
if(require('RUVnormalizeData')){
## Load the data
data('gender', package='RUVnormalizeData')
Y <- t(exprs(gender))
X <- as.numeric(phenoData(gender)$gender == 'M')
X <- X - mean(X)
X <- cbind(X/(sqrt(sum(X^2))))
chip <- annotation(gender)
## Extract regions and labs for plotting purposes
lregions <- sapply(rownames(Y),FUN=function(s) strsplit(s,'_')[[1]][2])
llabs <- sapply(rownames(Y),FUN=function(s) strsplit(s,'_')[[1]][3])
## Dimension of the factors
m <- nrow(Y)
n <- ncol(Y)
p <- ncol(X)
Y <- scale(Y, scale=FALSE) # Center gene expressions
cIdx <- which(featureData(gender)$isNegativeControl) # Negative control genes
## Prepare plots
annot <- cbind(as.character(sign(X)))
colnames(annot) <- 'gender'
plAnnots <- list('gender'='categorical')
lab.and.region <- apply(rbind(lregions, llabs),2,FUN=function(v) paste(v,collapse='_'))
gender.col <- c('-1' = "deeppink3", '1' = "blue")
## Remove platform effect by centering.
Y[chip=='hgu95a.db',] <- scale(Y[chip=='hgu95a.db',], scale=FALSE)
Y[chip=='hgu95av2.db',] <- scale(Y[chip=='hgu95av2.db',], scale=FALSE)
## Number of genes kept for clustering, based on their variance
nKeep <- 1260
##--------------------------
## Naive RUV-2 no shrinkage
##--------------------------
k <- 20
nu <- 0
## Correction
nsY <- naiveRandRUV(Y, cIdx, nu.coeff=0, k=k)
## Clustering of the corrected data
sdY <- apply(nsY, 2, sd)
ssd <- sort(sdY,decreasing=TRUE,index.return=TRUE)$ix
kmres2ns <- kmeans(nsY[,ssd[1:nKeep],drop=FALSE],centers=2,nstart=200)
vclust2ns <- kmres2ns$cluster
nsScore <- clScore(vclust2ns, X)
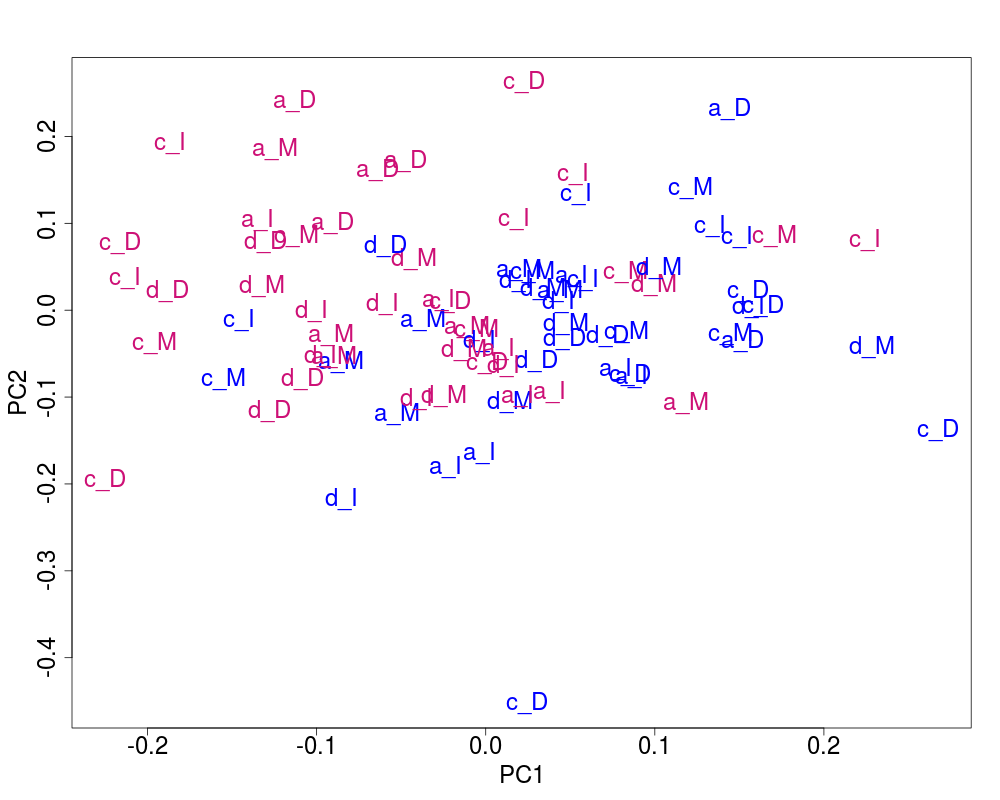
## Plot of the corrected data
svdRes2ns <- NULL
svdRes2ns <- svdPlot(nsY[, ssd[1:nKeep], drop=FALSE],
annot=annot,
labels=lab.and.region,
svdRes=svdRes2ns,
plAnnots=plAnnots,
kColors=gender.col, file=NULL)
##--------------------------
## Naive RUV-2 + shrinkage
##--------------------------
k <- m
nu.coeff <- 1e-2
## Correction
nY <- naiveRandRUV(Y, cIdx, nu.coeff=nu.coeff, k=k)
## Clustering of the corrected data
sdY <- apply(nY, 2, sd)
ssd <- sort(sdY,decreasing=TRUE,index.return=TRUE)$ix
kmres2 <- kmeans(nY[,ssd[1:nKeep],drop=FALSE],centers=2,nstart=200)
vclust2 <- kmres2$cluster
nScore <- clScore(vclust2,X)
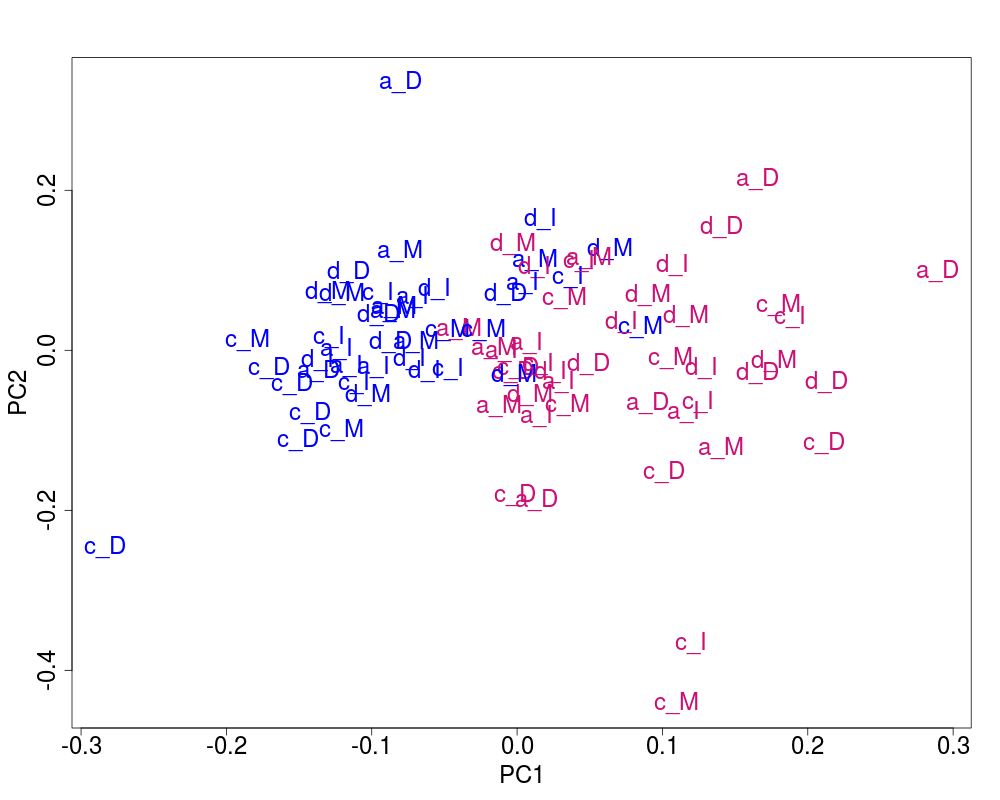
## Plot of the corrected data
svdRes2 <- NULL
svdRes2 <- svdPlot(nY[, ssd[1:nKeep], drop=FALSE],
annot=annot,
labels=lab.and.region,
svdRes=svdRes2,
plAnnots=plAnnots,
kColors=gender.col, file=NULL)
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(RUVnormalize)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/RUVnormalize/clScore.Rd_%03d_medium.png", width=480, height=480)
> ### Name: clScore
> ### Title: Computes a distance between two partitions of the same data
> ### Aliases: clScore
>
> ### ** Examples
>
> if(require('RUVnormalizeData')){
+
+ ## Load the data
+ data('gender', package='RUVnormalizeData')
+
+ Y <- t(exprs(gender))
+ X <- as.numeric(phenoData(gender)$gender == 'M')
+ X <- X - mean(X)
+ X <- cbind(X/(sqrt(sum(X^2))))
+ chip <- annotation(gender)
+
+ ## Extract regions and labs for plotting purposes
+ lregions <- sapply(rownames(Y),FUN=function(s) strsplit(s,'_')[[1]][2])
+ llabs <- sapply(rownames(Y),FUN=function(s) strsplit(s,'_')[[1]][3])
+
+ ## Dimension of the factors
+ m <- nrow(Y)
+ n <- ncol(Y)
+ p <- ncol(X)
+
+ Y <- scale(Y, scale=FALSE) # Center gene expressions
+
+ cIdx <- which(featureData(gender)$isNegativeControl) # Negative control genes
+
+ ## Prepare plots
+ annot <- cbind(as.character(sign(X)))
+ colnames(annot) <- 'gender'
+ plAnnots <- list('gender'='categorical')
+ lab.and.region <- apply(rbind(lregions, llabs),2,FUN=function(v) paste(v,collapse='_'))
+ gender.col <- c('-1' = "deeppink3", '1' = "blue")
+
+ ## Remove platform effect by centering.
+
+ Y[chip=='hgu95a.db',] <- scale(Y[chip=='hgu95a.db',], scale=FALSE)
+ Y[chip=='hgu95av2.db',] <- scale(Y[chip=='hgu95av2.db',], scale=FALSE)
+
+ ## Number of genes kept for clustering, based on their variance
+ nKeep <- 1260
+
+ ##--------------------------
+ ## Naive RUV-2 no shrinkage
+ ##--------------------------
+
+ k <- 20
+ nu <- 0
+
+ ## Correction
+ nsY <- naiveRandRUV(Y, cIdx, nu.coeff=0, k=k)
+
+ ## Clustering of the corrected data
+ sdY <- apply(nsY, 2, sd)
+ ssd <- sort(sdY,decreasing=TRUE,index.return=TRUE)$ix
+ kmres2ns <- kmeans(nsY[,ssd[1:nKeep],drop=FALSE],centers=2,nstart=200)
+ vclust2ns <- kmres2ns$cluster
+ nsScore <- clScore(vclust2ns, X)
+
+ ## Plot of the corrected data
+ svdRes2ns <- NULL
+ svdRes2ns <- svdPlot(nsY[, ssd[1:nKeep], drop=FALSE],
+ annot=annot,
+ labels=lab.and.region,
+ svdRes=svdRes2ns,
+ plAnnots=plAnnots,
+ kColors=gender.col, file=NULL)
+
+ ##--------------------------
+ ## Naive RUV-2 + shrinkage
+ ##--------------------------
+
+ k <- m
+ nu.coeff <- 1e-2
+
+ ## Correction
+ nY <- naiveRandRUV(Y, cIdx, nu.coeff=nu.coeff, k=k)
+
+ ## Clustering of the corrected data
+ sdY <- apply(nY, 2, sd)
+ ssd <- sort(sdY,decreasing=TRUE,index.return=TRUE)$ix
+ kmres2 <- kmeans(nY[,ssd[1:nKeep],drop=FALSE],centers=2,nstart=200)
+ vclust2 <- kmres2$cluster
+ nScore <- clScore(vclust2,X)
+
+ ## Plot of the corrected data
+ svdRes2 <- NULL
+ svdRes2 <- svdPlot(nY[, ssd[1:nKeep], drop=FALSE],
+ annot=annot,
+ labels=lab.and.region,
+ svdRes=svdRes2,
+ plAnnots=plAnnots,
+ kColors=gender.col, file=NULL)
+ }
Loading required package: RUVnormalizeData
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Warning message:
In naiveRandRUV(Y, cIdx, nu.coeff = nu.coeff, k = k) :
k larger than the rank of Y[, cIdx]. Using k=82 instead
>
>
>
>
>
> dev.off()
null device
1
>
 .
.