Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
PC-correlated SNPs in principal component analysisDescriptionTo calculate the SNP correlations between eigenvactors and SNP genotypes Usage
snpgdsPCACorr(pcaobj, gdsobj, snp.id=NULL, eig.which=NULL, num.thread=1L,
verbose=TRUE)
Arguments
ValueReturn a list:
Author(s)Xiuwen Zheng ReferencesPatterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genetics 2:e190. See Also
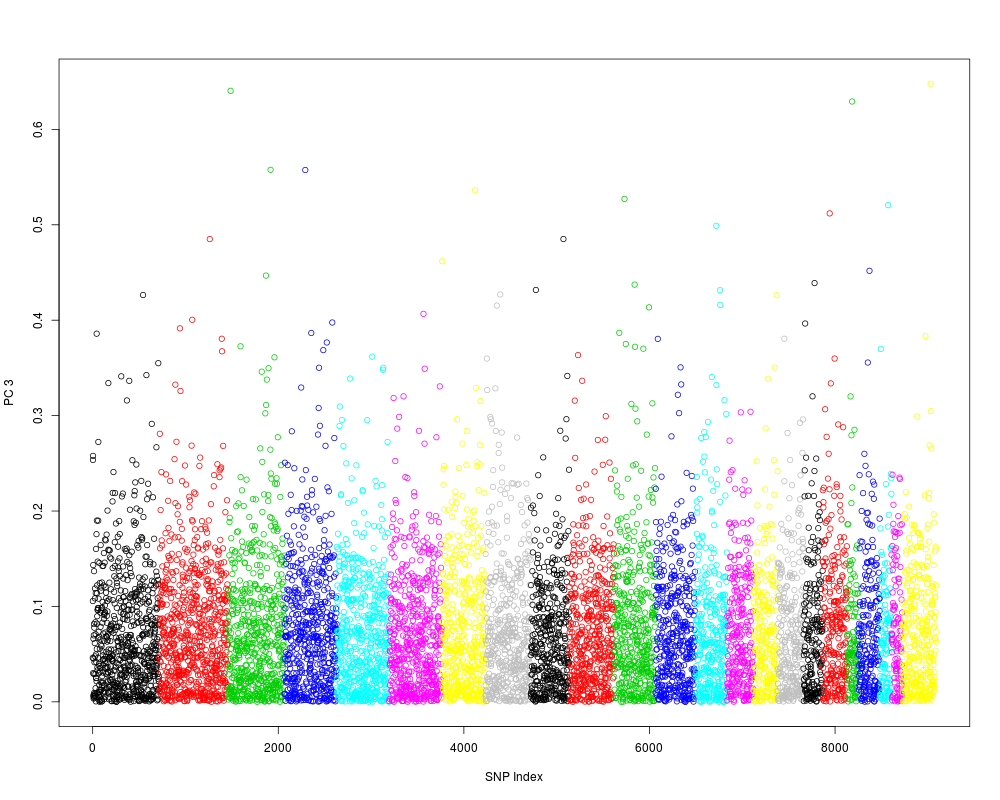
Examples# open an example dataset (HapMap) genofile <- snpgdsOpen(snpgdsExampleFileName()) # get chromosome index chr <- read.gdsn(index.gdsn(genofile, "snp.chromosome")) pca <- snpgdsPCA(genofile) CORR <- snpgdsPCACorr(pca, genofile, eig.which=1:4) plot(abs(CORR$snpcorr[3,]), xlab="SNP Index", ylab="PC 3", col=chr) # close the file snpgdsClose(genofile) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(SNPRelate)
Loading required package: gdsfmt
SNPRelate -- supported by Streaming SIMD Extensions 2 (SSE2)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/SNPRelate/snpgdsPCACorr.Rd_%03d_medium.png", width=480, height=480)
> ### Name: snpgdsPCACorr
> ### Title: PC-correlated SNPs in principal component analysis
> ### Aliases: snpgdsPCACorr
> ### Keywords: PCA GDS GWAS
>
> ### ** Examples
>
> # open an example dataset (HapMap)
> genofile <- snpgdsOpen(snpgdsExampleFileName())
> # get chromosome index
> chr <- read.gdsn(index.gdsn(genofile, "snp.chromosome"))
>
> pca <- snpgdsPCA(genofile)
Principal Component Analysis (PCA) on SNP genotypes:
Excluding 365 SNPs on non-autosomes
Excluding 1 SNP (monomorphic: TRUE, < MAF: NaN, or > missing rate: NaN)
Working space: 279 samples, 8722 SNPs
using 1 (CPU) core
PCA: the sum of all selected genotypes (0, 1 and 2) = 2446510
Wed Jul 6 05:34:47 2016 (internal increment: 1744)
[>.................................................] 0%, ETC: NA [==========>.......................................] 20%, ETC: 0s [====================>.............................] 40%, ETC: 0s [==============================>...................] 60%, ETC: 0s [========================================>.........] 80%, ETC: 0s [==================================================] 100%, ETC: 0s [==================================================] 100%, completed
Wed Jul 6 05:34:47 2016 Begin (eigenvalues and eigenvectors)
Wed Jul 6 05:34:47 2016 Done.
> CORR <- snpgdsPCACorr(pca, genofile, eig.which=1:4)
SNP correlation:
Working space: 279 samples, 9088 SNPs
Using 1 (CPU) core.
Using the top 32 eigenvectors.
SNP Correlation: the sum of all selected genotypes (0, 1 and 2) = 2553065
Wed Jul 6 05:34:47 2016 (internal increment: 13976)
[>.................................................] 0%, ETC: NA [==================================================] 100%, completed
Wed Jul 6 05:34:47 2016 Done.
> plot(abs(CORR$snpcorr[3,]), xlab="SNP Index", ylab="PC 3", col=chr)
>
> # close the file
> snpgdsClose(genofile)
>
>
>
>
>
> dev.off()
null device
1
>
|