Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
User friendly interface to class 'SampleSize'DescriptionUser friendly interface to class "SampleSize" Usage
sampleSize(PilotData,
method = c("deconv", "congrad", "tikhonov", "ferreira"),
control = list(from = -6, to = 6, resolution = 2^9))
Arguments
DetailsThe default method is 'deconv' which is an kernel deconvolution density estimator implementated using The 'control' argument is a list that can supply any of the following components. Per method logical checks are performed.
Valueobject of class SampleSize. Author(s)Maarten van Iterson Referencesvan Iterson, M., P. 't Hoen, P. Pedotti, G. Hooiveld, J. den Dunnen, G. van Ommen, J. Boer, and R. de Menezes (2009): 'Relative power and sample size analysis on gene expression profiling data,' BMC Genomics, 10, 439–449. Ferreira, J. and A. Zwinderman (2006a): 'Approximate Power and Sample Size Calculations with the Benjamini-Hochberg Method,' The International Journal of Biostatistics, 2, 1. Ferreira, J. and A. Zwinderman (2006b): 'Approximate Sample Size Calculations with Microarray Data: An Illustration,' Statistical Applications in Genetics and Molecular Biology, 5, 1. Hansen, P. (2010): Discrete Inverse Problems: Insight and Algorithms, SIAM: Fun- damentals of algorithms series. Langaas, M., B. Lindqvist, and E. Ferkingstad (2005): 'Estimating the proportion of true null hypotheses, with application to DNA microarray data,' Journal of the Royal Statistical Society Series B, 67, 555–572. Storey, J. (2003): 'The positive false discovery rate: A bayesian interpretation and the q-value,' Annals of Statistics, 31, 2013–2035. See Also
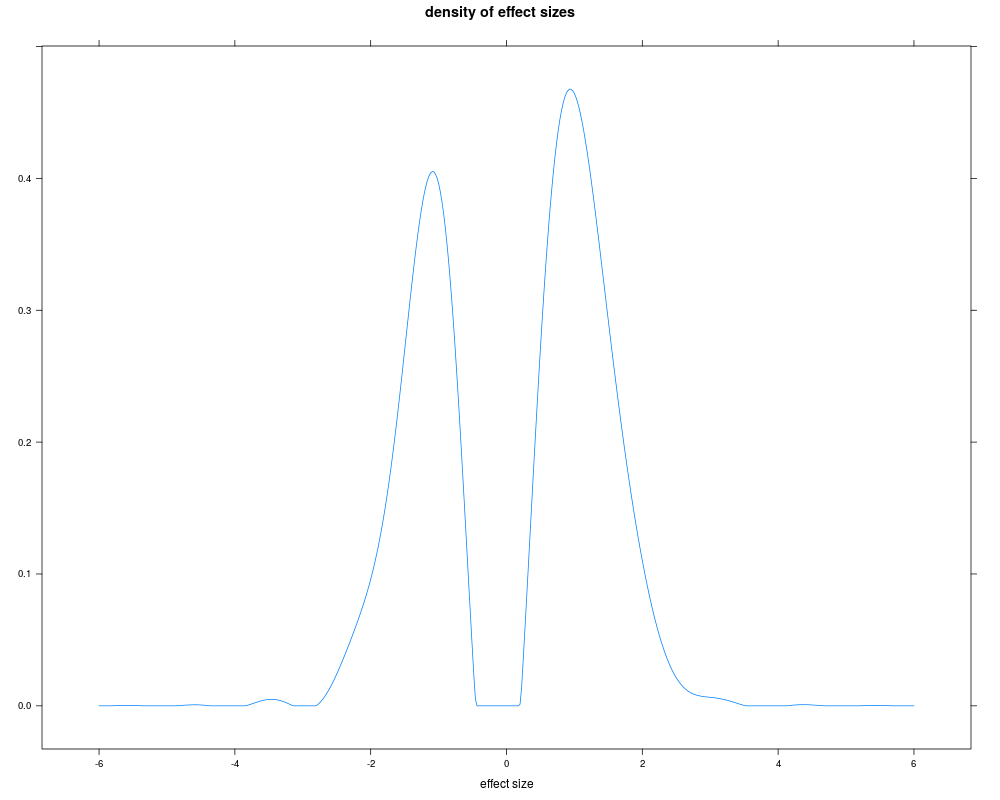
Examplesm <- 5000 ##number of genes J <- 10 ##sample size per group pi0 <- 0.8 ##proportion of non-differentially expressed genes m0 <- as.integer(m*pi0) mu <- rbitri(m - m0, a = log2(1.2), b = log2(4), m = log2(2)) #effect size distribution data <- simdat(mu, m=m, pi0=pi0, J=J, noise=NULL) library(genefilter) stat <- rowttests(data, factor(rep(c(0, 1), each=J)), tstatOnly=TRUE)$statistic pd <- pilotData(statistics=stat, samplesize=sqrt(J/2), distribution='norm') ss <- sampleSize(pd, method='deconv') plot(ss) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(SSPA)
Loading required package: qvalue
Loading required package: lattice
Loading required package: limma
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/SSPA/sampleSize.Rd_%03d_medium.png", width=480, height=480)
> ### Name: sampleSize
> ### Title: User friendly interface to class 'SampleSize'
> ### Aliases: sampleSize
>
> ### ** Examples
>
> m <- 5000 ##number of genes
> J <- 10 ##sample size per group
> pi0 <- 0.8 ##proportion of non-differentially expressed genes
> m0 <- as.integer(m*pi0)
> mu <- rbitri(m - m0, a = log2(1.2), b = log2(4), m = log2(2)) #effect size distribution
> data <- simdat(mu, m=m, pi0=pi0, J=J, noise=NULL)
> library(genefilter)
> stat <- rowttests(data, factor(rep(c(0, 1), each=J)), tstatOnly=TRUE)$statistic
> pd <- pilotData(statistics=stat, samplesize=sqrt(J/2), distribution='norm')
> ss <- sampleSize(pd, method='deconv')
> plot(ss)
>
>
>
>
>
> dev.off()
null device
1
>
|