Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Simulations to predict the number of loci expected in RAD and GBS approaches.DescriptionSimRAD provides a number of functions to simulate restriction enzyme digestion, library construction and fragments size selection to predict the number of loci expected from most of the Restriction site Associated DNA (RAD) and Genotyping By Sequencing (GBS) approaches. SimRAD aims to provide an estimation of the number of loci expected from a given genome depending on protocol type and parameters allowing to assess feasibility, multiplexing capacity and the amount of sequencing required. Details
This package provides a number of functions to simulate restriction enzyme digestion ( Author(s)Olivier Lepais and Jason Weir ReferencesLepais O & Weir JT. 2014. SimRAD: an R package for simulation-based prediction of the number of loci expected in RADseq and similar genotyping by sequencing approaches. Molecular Ecology Resources, 14, 1314-1321. DOI: 10.1111/1755-0998.12273. Examples
### Example 1: a single digestion (RAD)
simseq <- sim.DNAseq(size=100000, GCfreq=0.51)
#Restriction Enzyme 1
#SbfI
cs_5p1 <- "CCTGCA"
cs_3p1 <- "GG"
simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, verbose=TRUE)
### Example 2: a single digestion (GBS)
simseq <- sim.DNAseq(size=100000, GCfreq=0.51)
#Restriction Enzyme 1
# ApeKI : G|CWGC which is equivalent of either G|CAGC or G|CTGC
cs_5p1 <- "G"
cs_3p1 <- "CAGC"
cs_5p2 <- "G"
cs_3p2 <- "CTGC"
simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, cs_5p1, cs_3p1, verbose=TRUE)
### Example3: a double digestion (ddRAD)
# simulating some sequence:
simseq <- sim.DNAseq(size=1000000, GCfreq=0.433)
#Restriction Enzyme 1
#TaqI
cs_5p1 <- "T"
cs_3p1 <- "CGA"
#Restriction Enzyme 2
#MseI #
cs_5p2 <- "T"
cs_3p2 <- "TAA"
simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, cs_5p2, cs_3p2, verbose=TRUE)
simseq.sel <- adapt.select(simseq.dig, type="AB+BA", cs_5p1, cs_3p1, cs_5p2, cs_3p2)
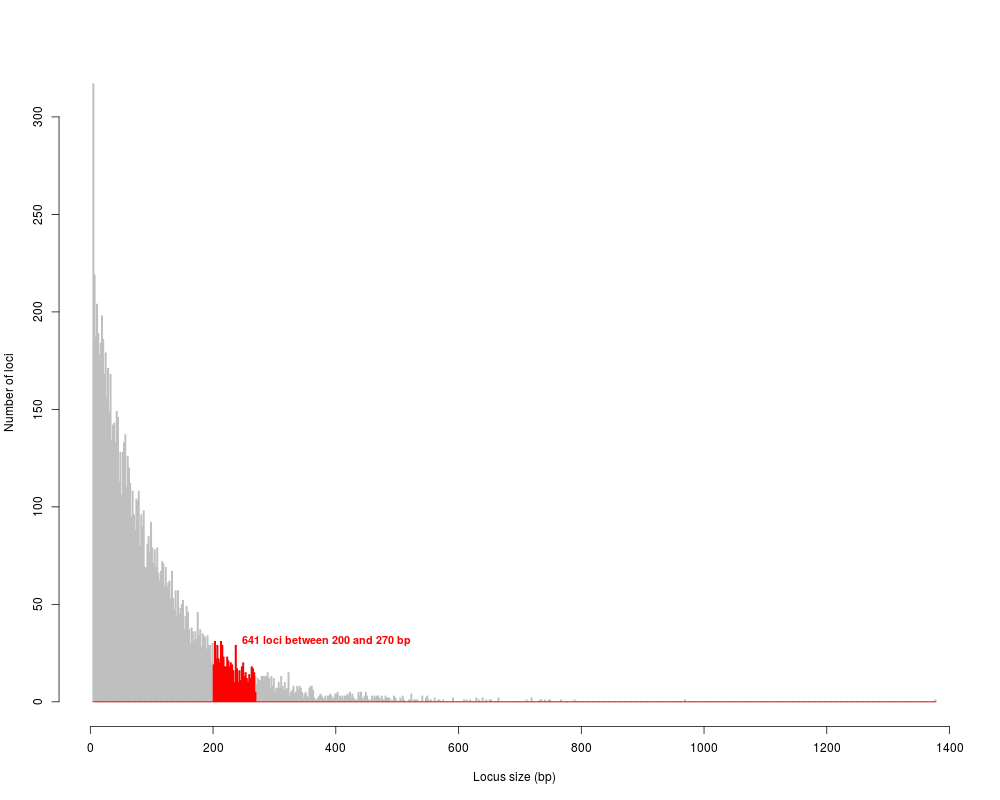
# wide size selection (200-270):
wid.simseq <- size.select(simseq.sel, min.size = 200, max.size = 270, graph=TRUE, verbose=TRUE)
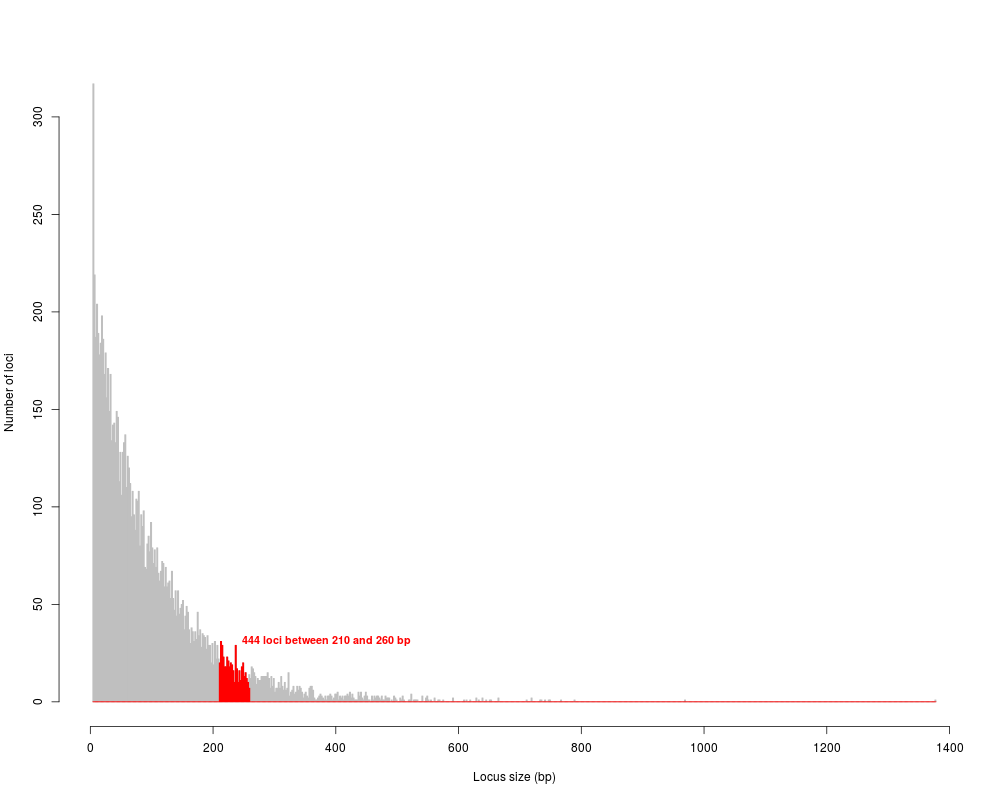
# narrow size selection (210-260):
nar.simseq <- size.select(simseq.sel, min.size = 210, max.size = 260, graph=TRUE, verbose=TRUE)
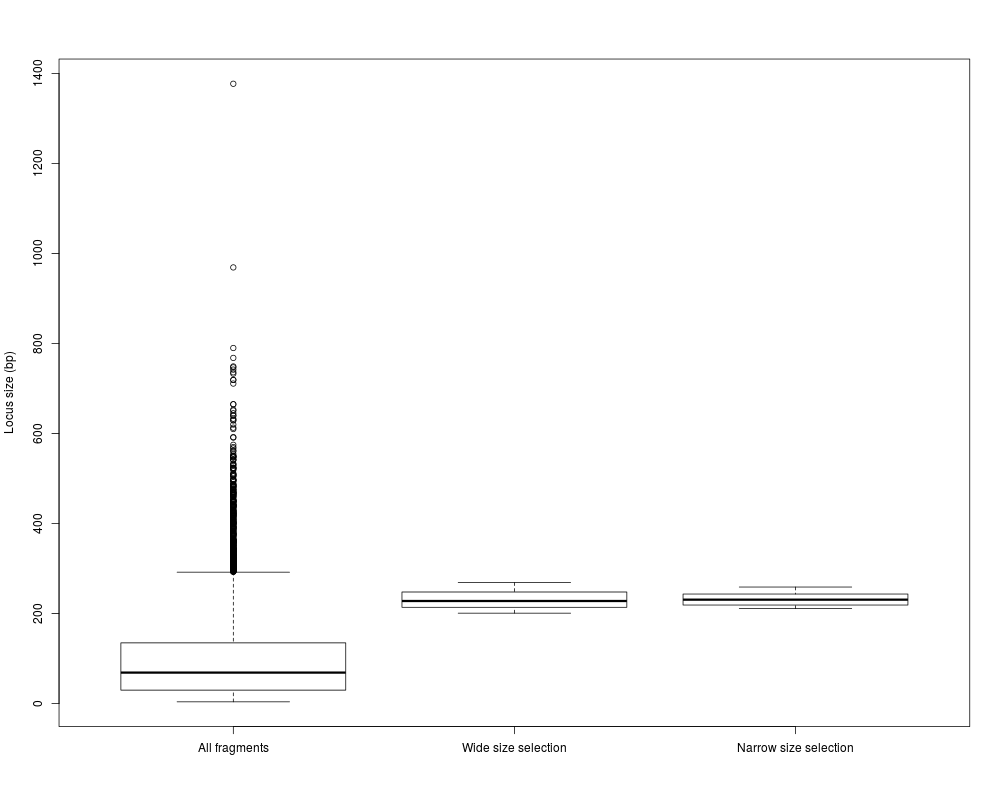
#the resulting fragment characteristics can be further examined:
boxplot(list(width(simseq.sel), width(wid.simseq), width(nar.simseq)), names=c("All fragments",
"Wide size selection", "Narrow size selection"), ylab="Locus size (bp)")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(SimRAD)
Loading required package: Biostrings
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Loading required package: ShortRead
Loading required package: BiocParallel
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: GenomicAlignments
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: zlibbioc
> png(filename="/home/ddbj/snapshot/RGM3/R_CC/result/SimRAD/SimRAD-package.Rd_%03d_medium.png", width=480, height=480)
> ### Name: SimRAD-package
> ### Title: Simulations to predict the number of loci expected in RAD and
> ### GBS approaches.
> ### Aliases: SimRAD-package SimRAD
> ### Keywords: package double digestion restriction enzyme library
> ### construction adaptator ligation fragment selection restriction
> ### exclusion RESTseq RAD GBS ddRAD ezRAD
>
> ### ** Examples
>
> ### Example 1: a single digestion (RAD)
> simseq <- sim.DNAseq(size=100000, GCfreq=0.51)
> #Restriction Enzyme 1
> #SbfI
> cs_5p1 <- "CCTGCA"
> cs_3p1 <- "GG"
> simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, verbose=TRUE)
Number of restriction sites: 4
>
> ### Example 2: a single digestion (GBS)
> simseq <- sim.DNAseq(size=100000, GCfreq=0.51)
> #Restriction Enzyme 1
> # ApeKI : G|CWGC which is equivalent of either G|CAGC or G|CTGC
> cs_5p1 <- "G"
> cs_3p1 <- "CAGC"
> cs_5p2 <- "G"
> cs_3p2 <- "CTGC"
> simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, cs_5p1, cs_3p1, verbose=TRUE)
Number of restriction sites for the first enzyme: 105
Number of restriction sites for the second enzyme: 105
Number of type AB and BA fragments:208
Number of type AA fragments:104
Number of type BB fragments:104
Warning messages:
1: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
2: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
3: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
4: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
>
> ### Example3: a double digestion (ddRAD)
> # simulating some sequence:
> simseq <- sim.DNAseq(size=1000000, GCfreq=0.433)
>
> #Restriction Enzyme 1
> #TaqI
> cs_5p1 <- "T"
> cs_3p1 <- "CGA"
> #Restriction Enzyme 2
> #MseI #
> cs_5p2 <- "T"
> cs_3p2 <- "TAA"
>
> simseq.dig <- insilico.digest(simseq, cs_5p1, cs_3p1, cs_5p2, cs_3p2, verbose=TRUE)
Number of restriction sites for the first enzyme: 3708
Number of restriction sites for the second enzyme: 6451
Number of type AB and BA fragments:10158
Number of type AA fragments:3708
Number of type BB fragments:6450
Warning messages:
1: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
2: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
3: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
4: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
> simseq.sel <- adapt.select(simseq.dig, type="AB+BA", cs_5p1, cs_3p1, cs_5p2, cs_3p2)
Warning messages:
1: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
2: In NSBS(i, x, exact = exact, upperBoundIsStrict = !allow.append) :
subscript is an array, passing it thru as.vector() first
>
> # wide size selection (200-270):
> wid.simseq <- size.select(simseq.sel, min.size = 200, max.size = 270, graph=TRUE, verbose=TRUE)
651 fragments between 200 and 270 bp
>
> # narrow size selection (210-260):
> nar.simseq <- size.select(simseq.sel, min.size = 210, max.size = 260, graph=TRUE, verbose=TRUE)
450 fragments between 210 and 260 bp
>
> #the resulting fragment characteristics can be further examined:
> boxplot(list(width(simseq.sel), width(wid.simseq), width(nar.simseq)), names=c("All fragments",
+ "Wide size selection", "Narrow size selection"), ylab="Locus size (bp)")
>
>
>
>
>
>
> dev.off()
null device
1
>
|