R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(Sushi)
Loading required package: zoo
Attaching package: 'zoo'
The following objects are masked from 'package:base':
as.Date, as.Date.numeric
Loading required package: biomaRt
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/Sushi/plotBedgraph.Rd_%03d_medium.png", width=480, height=480)
> ### Name: plotBedgraph
> ### Title: plots data stored in bed file format
> ### Aliases: plotBedgraph
>
> ### ** Examples
>
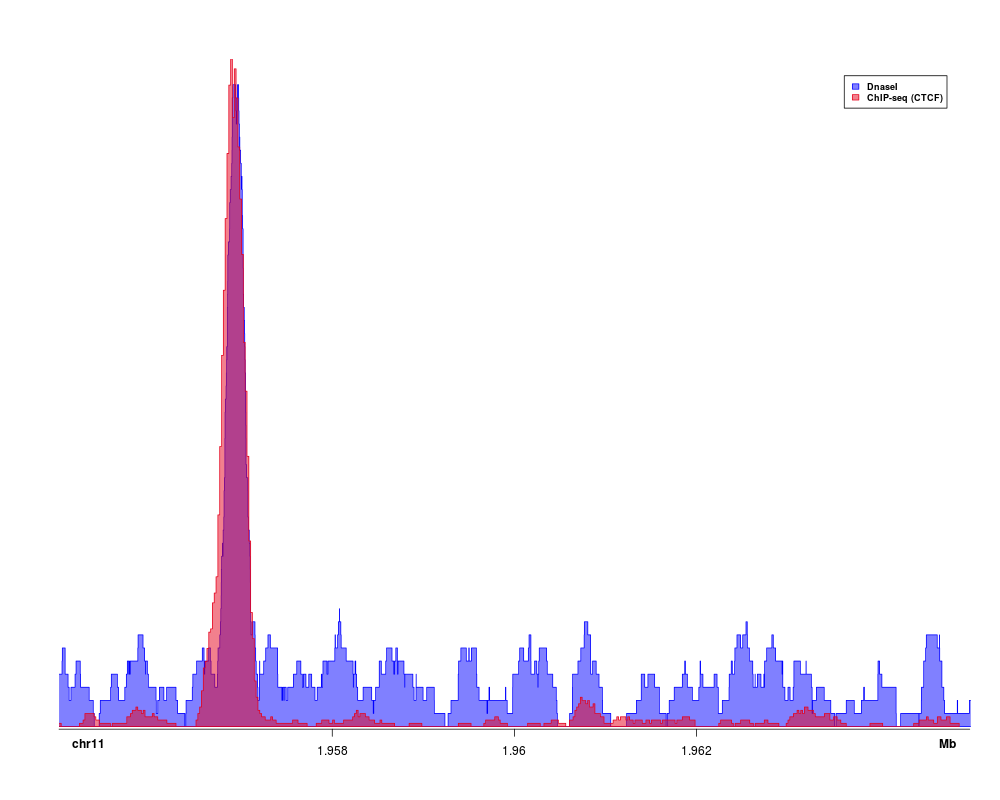
> data(Sushi_ChIPSeq_CTCF.bedgraph)
> data(Sushi_DNaseI.bedgraph)
>
> chrom = "chr11"
> chromstart = 1955000
> chromend = 1965000
>
> plotBedgraph(Sushi_ChIPSeq_CTCF.bedgraph,chrom,chromstart,chromend,transparency=.50,flip=FALSE,color="blue",linecol="blue")
> plotBedgraph(Sushi_DNaseI.bedgraph,chrom,chromstart,chromend,transparency=.50,flip=FALSE,color="#E5001B",linecol="#E5001B",overlay=TRUE,rescaleoverlay=TRUE)
> labelgenome(chrom,chromstart,chromend,side=1,scipen=20,n=3,line=.18,chromline=.5,scaleline=0.5,scale="Mb")
>
> transparency = 0.5
> col1 = col2rgb("blue")
> finalcolor1 = rgb(col1[1],col1[2],col1[3],alpha=transparency * 255,maxColorValue = 255)
> col2 = col2rgb("#E5001B")
> finalcolor2 = rgb(col2[1],col2[2],col2[3],alpha=transparency * 255,maxColorValue = 255)
>
> legend("topright",inset=0.025,legend=c("DnaseI","ChIP-seq (CTCF)"),fill=c(finalcolor1,finalcolor2),border=c("blue","#E5001B"),text.font=2,cex=0.75)
>
>
>
>
>
> dev.off()
null device
1
>
 .
.