Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
A container for storing information used in TCCDescriptionThis is a container class for TCC. This class initially contains count data matrix and some information for the analysis of count data. It also provides further fields that are populated during the analysis. DetailsThis class is implemented as an R5 reference class.
Functions calling such methods copies the object prior to
calling the method to keep the semantics of functional programming.
This class can be created by the generic The values (defaults to all 1) in the FieldsThis class contains the following fields:
Examples
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2, DEG.assign = c(0.8, 0.2),
DEG.foldchange = c(4, 4), replicates = c(3, 3))
# Check the TCC class object.
tcc
# Check the fields of TCC class object.
names(tcc)
head(tcc$count)
# Check the normalization factors.
tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
iteration = 1, FDR = 0.1, floorPDEG = 0.05)
tcc$norm.factors
# Check the p-values and q-values.
tcc <- estimateDE(tcc, test.method = "edger", FDR = 0.1)
tcc
# Compare the breakdowns of estimated DEGs with the truth.
head(tcc$estimatedDEG)
head(tcc$simulation$trueDEG)
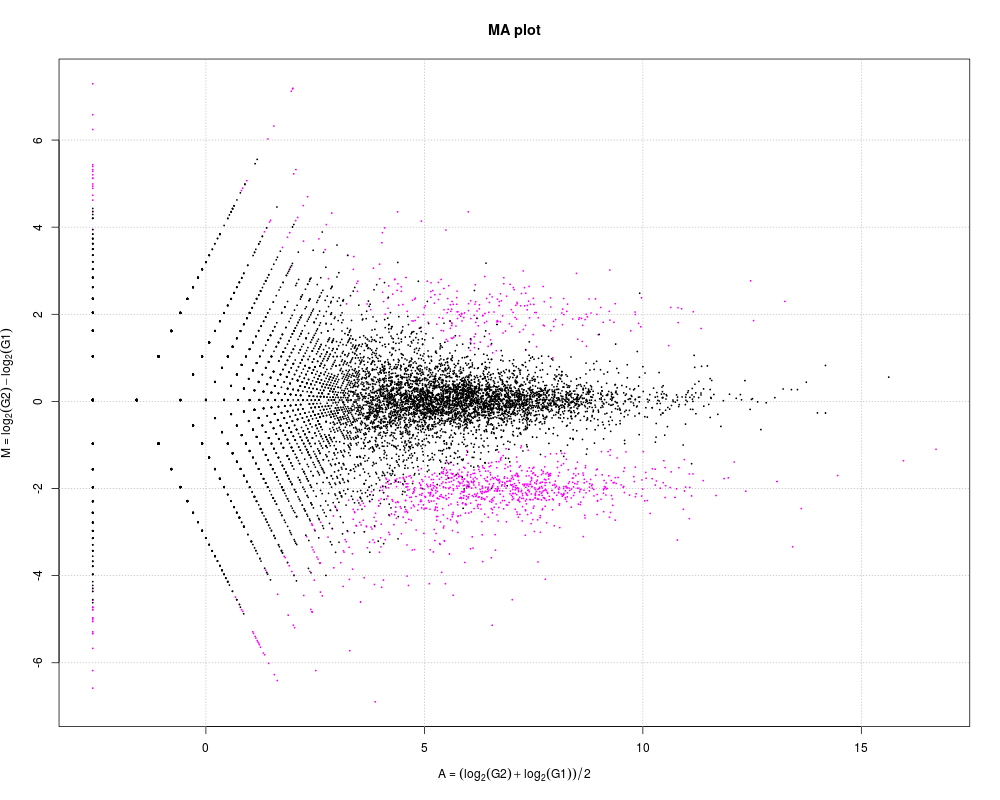
# M-A plotting.
plot(tcc)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TCC)
Loading required package: DESeq
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: locfit
locfit 1.5-9.1 2013-03-22
Loading required package: lattice
Welcome to 'DESeq'. For improved performance, usability and
functionality, please consider migrating to 'DESeq2'.
Loading required package: DESeq2
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Attaching package: 'DESeq2'
The following objects are masked from 'package:DESeq':
estimateSizeFactorsForMatrix, getVarianceStabilizedData,
varianceStabilizingTransformation
Loading required package: edgeR
Loading required package: limma
Attaching package: 'limma'
The following object is masked from 'package:DESeq2':
plotMA
The following object is masked from 'package:DESeq':
plotMA
The following object is masked from 'package:BiocGenerics':
plotMA
Loading required package: baySeq
Loading required package: abind
Loading required package: perm
Loading required package: ROC
Attaching package: 'TCC'
The following object is masked from 'package:edgeR':
calcNormFactors
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TCC/TCC-class.Rd_%03d_medium.png", width=480, height=480)
> ### Name: TCC-class
> ### Title: A container for storing information used in TCC
> ### Aliases: TCC-class show show.TCC show,TCC-method names names,TCC-method
> ### length length,TCC-method [ [,TCC,ANY-method [,TCC-method
> ### [,TCC,ANY,ANY-method [,TCC,ANY,ANY,ANY-method subset
> ### subset,TCC-method
> ### Keywords: classes
>
> ### ** Examples
>
> tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2, DEG.assign = c(0.8, 0.2),
+ DEG.foldchange = c(4, 4), replicates = c(3, 3))
TCC::INFO: Generating simulation data under NB distribution ...
TCC::INFO: (genesizes : 10000 )
TCC::INFO: (replicates : 3, 3 )
TCC::INFO: (PDEG : 0.16, 0.04 )
>
> # Check the TCC class object.
> tcc
Count:
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
gene_1 480 323 603 146 119 112
gene_2 126 114 125 41 32 59
gene_3 12 3 9 0 2 1
gene_4 575 593 1302 222 64 307
gene_5 52 54 41 11 5 6
gene_6 213 264 400 90 107 63
Sample:
group norm.factors lib.sizes
G1_rep1 1 1 1286214
G1_rep2 1 1 1288977
G1_rep3 1 1 1358796
G2_rep1 2 1 1052820
G2_rep2 2 1 1100227
G2_rep3 2 1 1039522
>
> # Check the fields of TCC class object.
> names(tcc)
[1] "count" "gene_id" "group" "norm.factors" "DEGES"
[6] "stat" "estimatedDEG" "simulation"
> head(tcc$count)
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
gene_1 480 323 603 146 119 112
gene_2 126 114 125 41 32 59
gene_3 12 3 9 0 2 1
gene_4 575 593 1302 222 64 307
gene_5 52 54 41 11 5 6
gene_6 213 264 400 90 107 63
>
> # Check the normalization factors.
> tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
+ iteration = 1, FDR = 0.1, floorPDEG = 0.05)
TCC::INFO: Calculating normalization factors using DEGES
TCC::INFO: (iDEGES pipeline : tmm - [ edger - tmm ] X 1 )
TCC::INFO: Done.
> tcc$norm.factors
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
0.9236479 0.9177329 0.8724595 1.1052712 1.0578330 1.1230555
>
> # Check the p-values and q-values.
> tcc <- estimateDE(tcc, test.method = "edger", FDR = 0.1)
TCC::INFO: Identifying DE genes using edger ...
TCC::INFO: Done.
> tcc
Count:
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
gene_1 480 323 603 146 119 112
gene_2 126 114 125 41 32 59
gene_3 12 3 9 0 2 1
gene_4 575 593 1302 222 64 307
gene_5 52 54 41 11 5 6
gene_6 213 264 400 90 107 63
Sample:
group norm.factors lib.sizes
G1_rep1 1 0.9236479 1188009
G1_rep2 1 0.9177329 1182937
G1_rep3 1 0.8724595 1185494
G2_rep1 2 1.1052712 1163652
G2_rep2 2 1.0578330 1163856
G2_rep3 2 1.1230555 1167441
DEGES:
Pipeline : tmm - [ edger - tmm ] X 1
Execution time : 2.5 sec
Threshold type : FDR < 0.10
Potential PDEG : 0.13
Results:
gene_id a.value m.value p.value q.value rank estimatedDEG
1 gene_1 7.922910 -1.873277 5.316356e-06 0.0001158247 459 1
2 gene_2 6.192864 -1.442605 1.133242e-03 0.0114584632 989 1
3 gene_3 1.499437 -2.973764 3.388453e-02 0.2163763378 1566 0
4 gene_4 8.655792 -2.034069 2.570919e-04 0.0030789451 835 1
5 gene_5 4.244826 -2.714812 5.345076e-05 0.0007825880 683 1
6 gene_6 7.314788 -1.728673 6.260202e-05 0.0008981638 697 1
>
> # Compare the breakdowns of estimated DEGs with the truth.
> head(tcc$estimatedDEG)
[1] 1 1 0 1 1 1
> head(tcc$simulation$trueDEG)
gene_1 gene_2 gene_3 gene_4 gene_5 gene_6
1 1 1 1 1 1
>
> # M-A plotting.
> plot(tcc)
>
>
>
>
>
> dev.off()
null device
1
>
|