Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Plot a log fold-change versus log average expression (so-called M-A plot)DescriptionThis function generates a scatter plot of log fold-change (i.e., M = log2(G2) - log2(G1) on the y-axis between Groups 1 vs. 2) versus log average expression (i.e., A = (log2(G1) + log2(G2)) / 2 on the x-axis) using normalized count data. Usage
## S3 method for class 'TCC'
plot(x, FDR = NULL, median.lines = FALSE, floor = 0,
group = NULL, col = NULL, col.tag = NULL, normalize = TRUE, ...)
Arguments
DetailsThis function generates roughly three different M-A plots
depending on the conditions for TCC-class objects.
When the function is performed just after the Genes with normalized counts of 0 in any one group cannot be plotted on the M-A plot because those M and A values cannot be calculated (as log 0 is undefined). Those points are plotted at the left side of the M-A plot, depending on the minimum A (i.e., log average expression) value. The x coordinate of those points is the minimum A value minus one. The y coordinate is calculated as if the zero count was the minimum observed non zero count in each group. ValueA scatter plot to the current graphic device. Examples
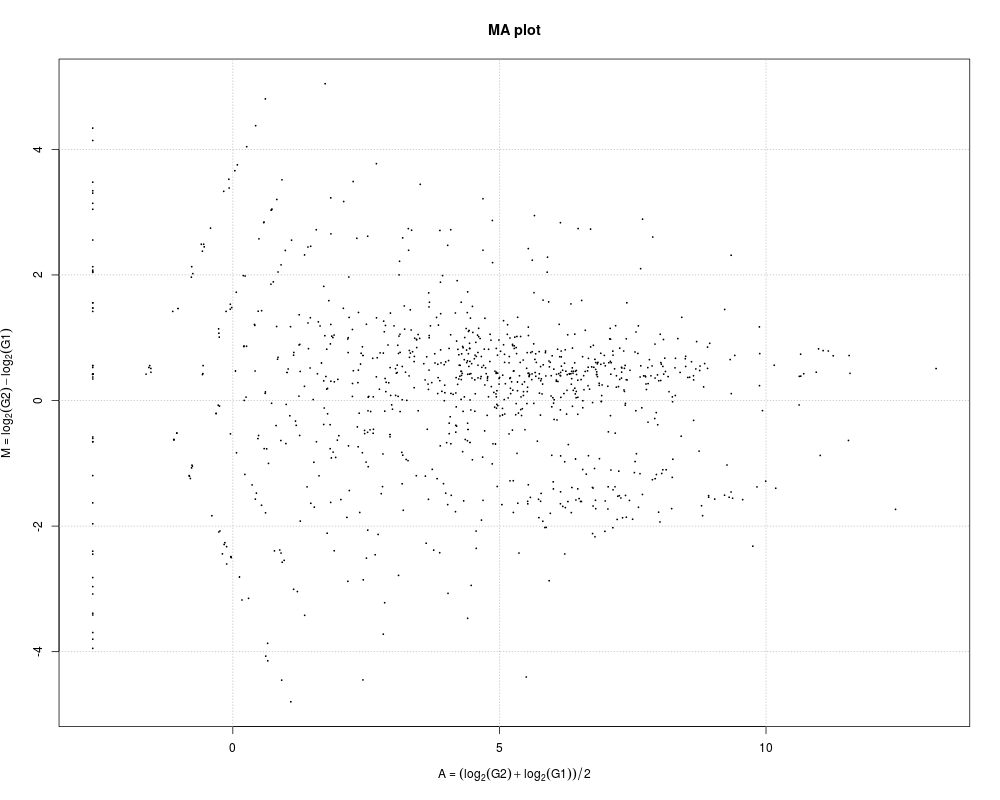
# Example 1.
# M-A plotting just after constructing the TCC class object from
# hypoData. In this case, the plot is generated from hypoData
# that has been scaled in such a way that the library sizes of
# each sample are the same as the mean library size of the
# original hypoData. Note that all points are in black. This is
# because the information about DEG or non-DEG for each gene is
# not indicated.
data(hypoData)
group <- c(1, 1, 1, 2, 2, 2)
tcc <- new("TCC", hypoData, group)
plot(tcc)
normalized.count <- getNormalizedData(tcc)
colSums(normalized.count)
colSums(hypoData)
mean(colSums(hypoData))
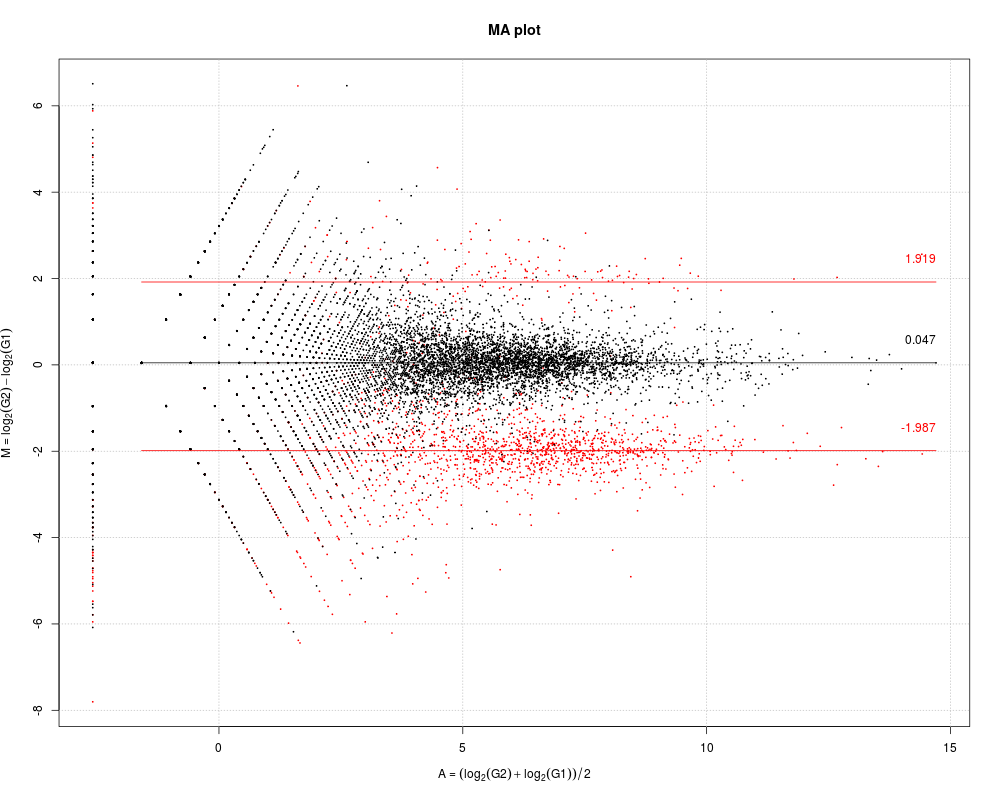
# Example 2.
# M-A plotting of DEGES/edgeR-normalized simulation data.
# It can be seen that the median M value for non-DEGs approaches
# zero. Note that non-DEGs are in black, DEGs are in red.
tcc <- simulateReadCounts()
tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
iteration = 1, FDR = 0.1, floorPDEG = 0.05)
plot(tcc, median.lines = TRUE)
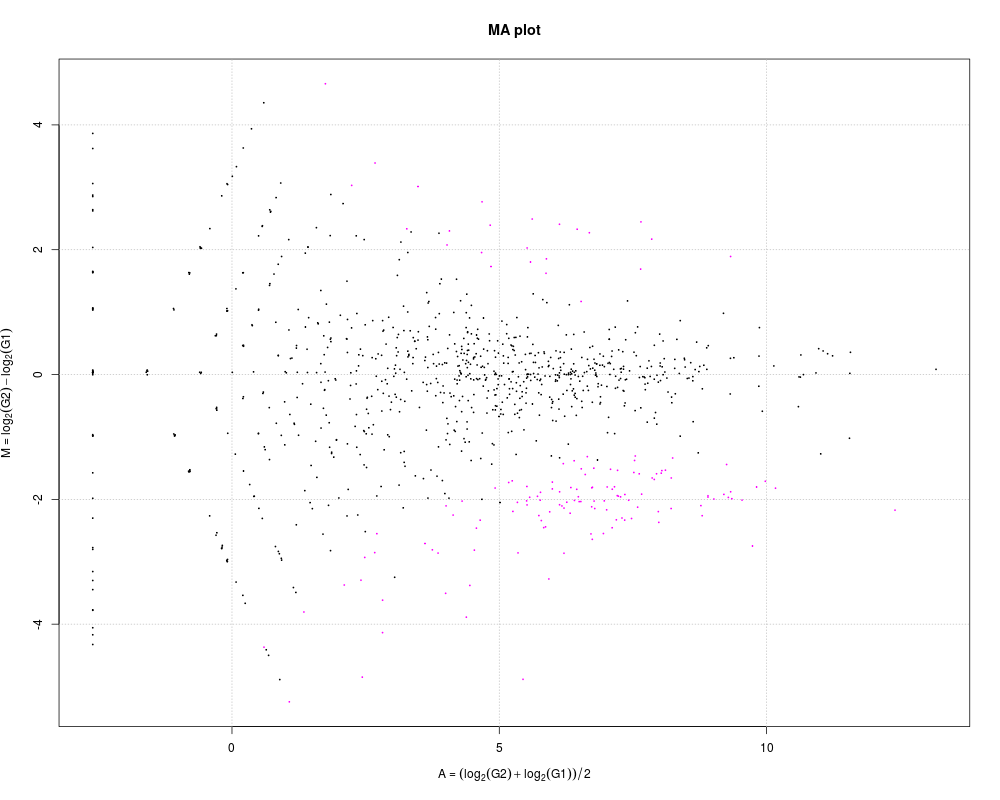
# Example 3.
# M-A plotting of DEGES/edgeR-normalized hypoData after performing
# DE analysis.
data(hypoData)
group <- c(1, 1, 1, 2, 2, 2)
tcc <- new("TCC", hypoData, group)
tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
iteration = 1, FDR = 0.1, floorPDEG = 0.05)
tcc <- estimateDE(tcc, test.method = "edger", FDR = 0.1)
plot(tcc)
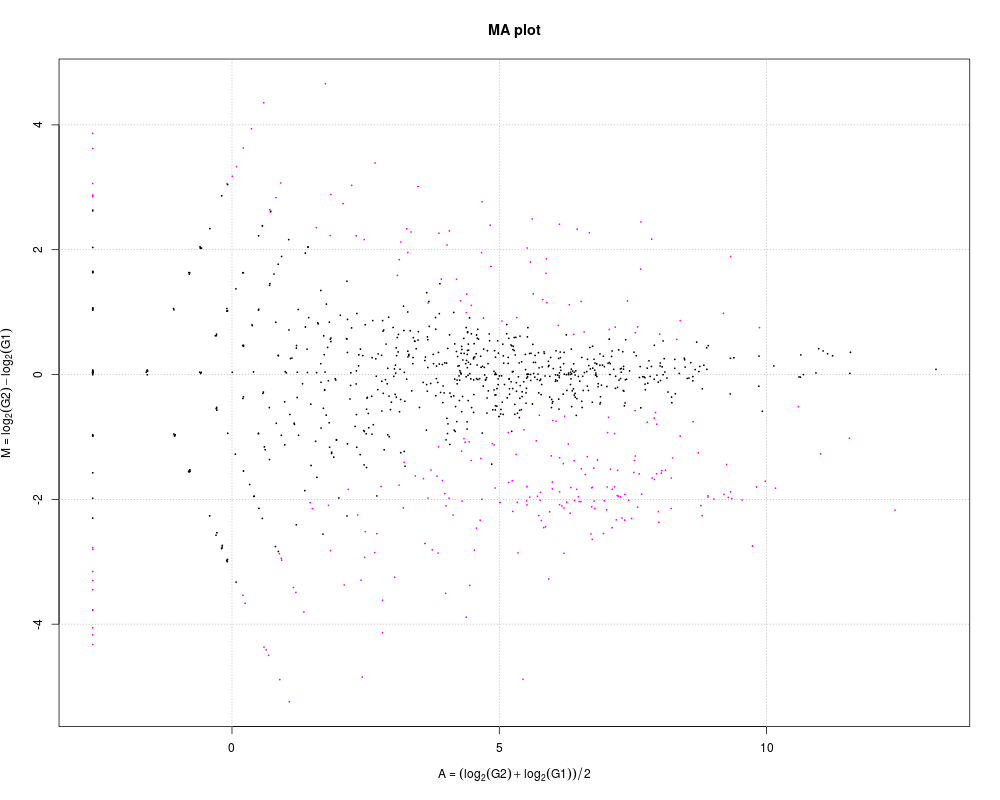
# Changing the FDR threshold
plot(tcc, FDR = 0.7)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TCC)
Loading required package: DESeq
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: locfit
locfit 1.5-9.1 2013-03-22
Loading required package: lattice
Welcome to 'DESeq'. For improved performance, usability and
functionality, please consider migrating to 'DESeq2'.
Loading required package: DESeq2
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Attaching package: 'DESeq2'
The following objects are masked from 'package:DESeq':
estimateSizeFactorsForMatrix, getVarianceStabilizedData,
varianceStabilizingTransformation
Loading required package: edgeR
Loading required package: limma
Attaching package: 'limma'
The following object is masked from 'package:DESeq2':
plotMA
The following object is masked from 'package:DESeq':
plotMA
The following object is masked from 'package:BiocGenerics':
plotMA
Loading required package: baySeq
Loading required package: abind
Loading required package: perm
Loading required package: ROC
Attaching package: 'TCC'
The following object is masked from 'package:edgeR':
calcNormFactors
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TCC/plot.TCC.Rd_%03d_medium.png", width=480, height=480)
> ### Name: plot
> ### Title: Plot a log fold-change versus log average expression (so-called
> ### M-A plot)
> ### Aliases: plot.TCC plot
> ### Keywords: methods
>
> ### ** Examples
>
> # Example 1.
> # M-A plotting just after constructing the TCC class object from
> # hypoData. In this case, the plot is generated from hypoData
> # that has been scaled in such a way that the library sizes of
> # each sample are the same as the mean library size of the
> # original hypoData. Note that all points are in black. This is
> # because the information about DEG or non-DEG for each gene is
> # not indicated.
> data(hypoData)
> group <- c(1, 1, 1, 2, 2, 2)
> tcc <- new("TCC", hypoData, group)
> plot(tcc)
>
> normalized.count <- getNormalizedData(tcc)
> colSums(normalized.count)
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
125922.3 125922.3 125922.3 125922.3 125922.3 125922.3
> colSums(hypoData)
G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3
142177 145289 149886 112100 104107 101975
> mean(colSums(hypoData))
[1] 125922.3
>
>
> # Example 2.
> # M-A plotting of DEGES/edgeR-normalized simulation data.
> # It can be seen that the median M value for non-DEGs approaches
> # zero. Note that non-DEGs are in black, DEGs are in red.
> tcc <- simulateReadCounts()
TCC::INFO: Generating simulation data under NB distribution ...
TCC::INFO: (genesizes : 10000 )
TCC::INFO: (replicates : 3, 3 )
TCC::INFO: (PDEG : 0.18, 0.02 )
> tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
+ iteration = 1, FDR = 0.1, floorPDEG = 0.05)
TCC::INFO: Calculating normalization factors using DEGES
TCC::INFO: (iDEGES pipeline : tmm - [ edger - tmm ] X 1 )
TCC::INFO: Done.
> plot(tcc, median.lines = TRUE)
>
>
> # Example 3.
> # M-A plotting of DEGES/edgeR-normalized hypoData after performing
> # DE analysis.
> data(hypoData)
> group <- c(1, 1, 1, 2, 2, 2)
> tcc <- new("TCC", hypoData, group)
> tcc <- calcNormFactors(tcc, norm.method = "tmm", test.method = "edger",
+ iteration = 1, FDR = 0.1, floorPDEG = 0.05)
TCC::INFO: Calculating normalization factors using DEGES
TCC::INFO: (iDEGES pipeline : tmm - [ edger - tmm ] X 1 )
TCC::INFO: Done.
> tcc <- estimateDE(tcc, test.method = "edger", FDR = 0.1)
TCC::INFO: Identifying DE genes using edger ...
TCC::INFO: Done.
> plot(tcc)
>
> # Changing the FDR threshold
> plot(tcc, FDR = 0.7)
>
>
>
>
>
> dev.off()
null device
1
>
|