Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
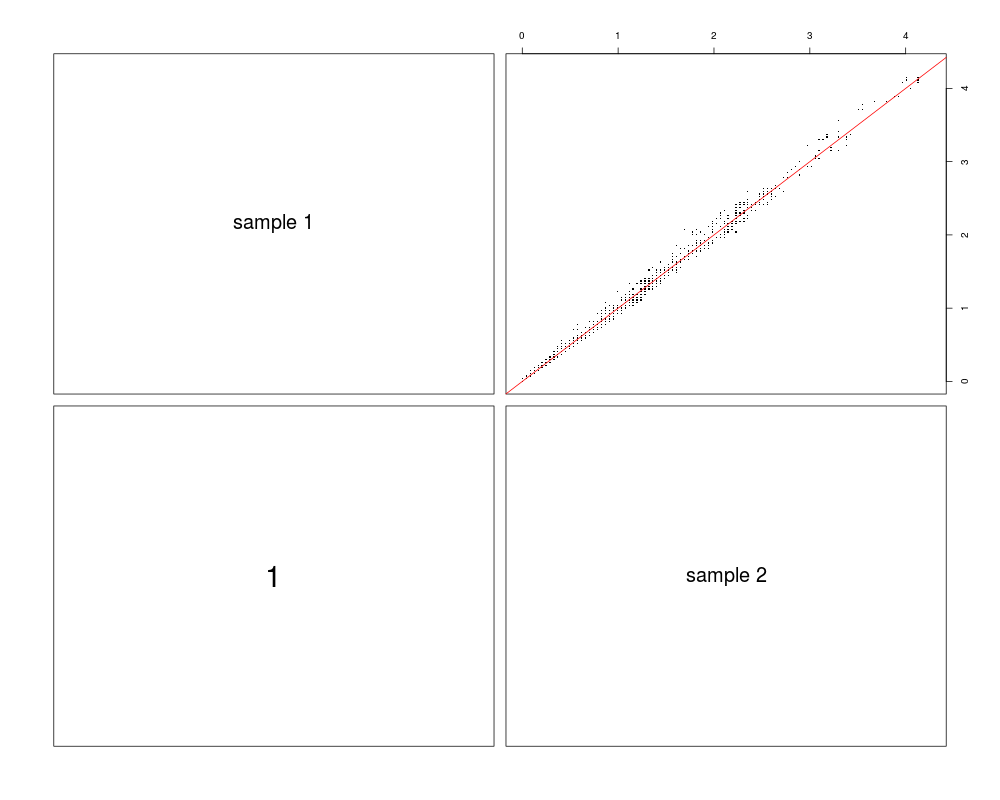
Coverage correlation plotDescriptionVisualization of target coverage correlations between pairs of samples. Usagecoverage.correlation(coveragelist, normalized = TRUE, plotfrac = 0.001, seed = 123, labels, main, pch = ".", cex.labels,
cex.pch = 2, cex.main = 1.2, cex.corr, font.labels = 1, font.main = 2, ...)
Arguments
DetailsIf Value'pairs'-style plot where upper panels show scatter plot of (a randomly chosen fraction of) coverage values for pairs of samples. The lower panels show the respective Pearson correlation coefficients, calculated using all coverage values (even if not all of them are shown in the scatter plot). Author(s)Manuela Hummel m.hummel@dkfz.de See Also
Examples
## get reads and targets
exptPath <- system.file("extdata", package="TEQC")
readsfile <- file.path(exptPath, "ExampleSet_Reads.bed")
reads <- get.reads(readsfile, idcol=4, skip=0)
targetsfile <- file.path(exptPath, "ExampleSet_Targets.bed")
targets <- get.targets(targetsfile, skip=0)
## calculate per-base coverages
Coverage <- coverage.target(reads, targets, perBase=TRUE)
## simulate another sample
r <- sample(nrow(reads), 0.1 * nrow(reads))
reads2 <- reads[-r,,drop=TRUE]
Coverage2 <- coverage.target(reads2, targets, perBase=TRUE)
## coverage uniformity plot
covlist <- list(Coverage, Coverage2)
coverage.correlation(covlist, plotfrac=0.1)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TEQC)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: IRanges
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: Biostrings
Loading required package: XVector
Loading required package: hwriter
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TEQC/coverage.correlation.Rd_%03d_medium.png", width=480, height=480)
> ### Name: coverage.correlation
> ### Title: Coverage correlation plot
> ### Aliases: coverage.correlation
> ### Keywords: hplot
>
> ### ** Examples
>
> ## get reads and targets
> exptPath <- system.file("extdata", package="TEQC")
> readsfile <- file.path(exptPath, "ExampleSet_Reads.bed")
> reads <- get.reads(readsfile, idcol=4, skip=0)
[1] "read 19546 sequenced reads"
> targetsfile <- file.path(exptPath, "ExampleSet_Targets.bed")
> targets <- get.targets(targetsfile, skip=0)
[1] "read 50 (non-overlapping) target regions"
Warning message:
the "reduce" method for RangedData object is deprecated
>
> ## calculate per-base coverages
> Coverage <- coverage.target(reads, targets, perBase=TRUE)
>
> ## simulate another sample
> r <- sample(nrow(reads), 0.1 * nrow(reads))
> reads2 <- reads[-r,,drop=TRUE]
> Coverage2 <- coverage.target(reads2, targets, perBase=TRUE)
>
> ## coverage uniformity plot
> covlist <- list(Coverage, Coverage2)
> coverage.correlation(covlist, plotfrac=0.1)
>
>
>
>
>
> dev.off()
null device
1
>
|