Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
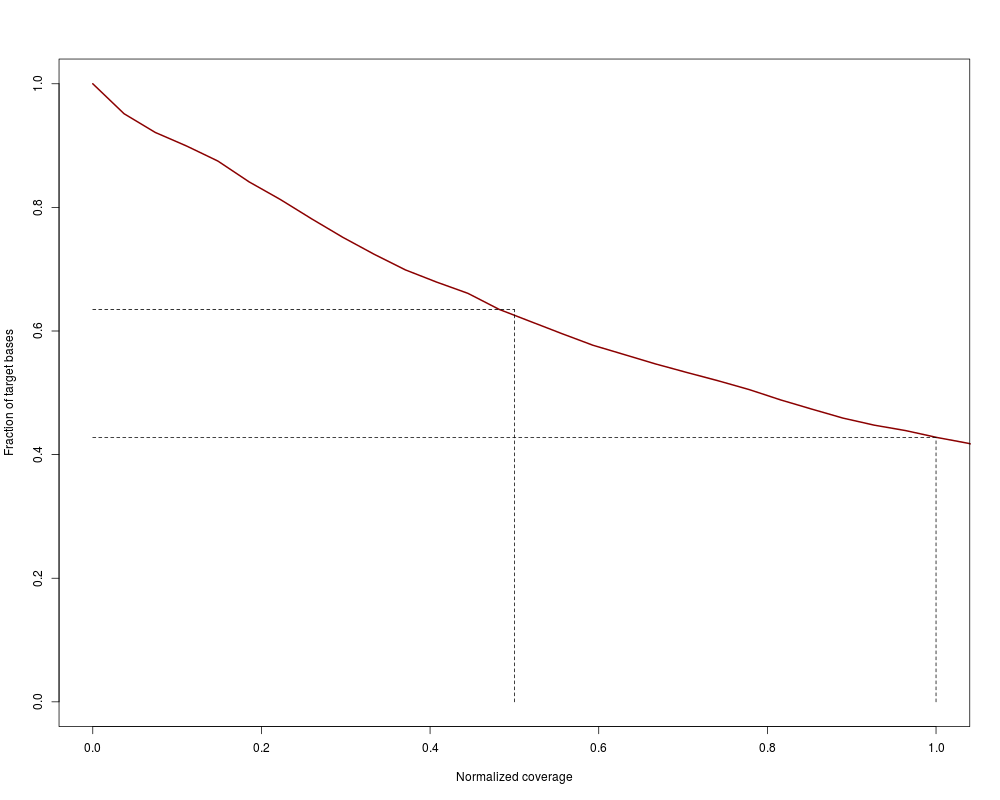
Coverage uniformity plotDescriptionVisualization of target coverage uniformity. A line shows the cumulative fraction of targeted bases that reach at least a certain normalized coverage. Usagecoverage.uniformity(coveragelist, addlines = TRUE, add = FALSE, xlab, ylab, xlim, ylim, col, lwd, ...) Arguments
DetailsThe function calculates normalized coverages: per-base coverages divided by average coverage over all targeted bases. Normalized coverages are not dependent on the absolute quantity of reads and are hence better comparable between different samples or even different experiments. ValueLine plot showing the fraction of targeted bases (y-axis) achieving a normalized
coverage of at least x. The x-axis by default is truncated at 1, which corresponds to the average
normalized coverage. The steeper the curve is falling, the less uniform is the coverage.
If Author(s)Manuela Hummel m.hummel@dkfz.de ReferencesGnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009; 27(2): 182-9. See Also
Examples
## get reads and targets
exptPath <- system.file("extdata", package="TEQC")
readsfile <- file.path(exptPath, "ExampleSet_Reads.bed")
reads <- get.reads(readsfile, idcol=4, skip=0)
targetsfile <- file.path(exptPath, "ExampleSet_Targets.bed")
targets <- get.targets(targetsfile, skip=0)
## calculate per-base coverages
Coverage <- coverage.target(reads, targets, perBase=TRUE)
## coverage uniformity plot
coverage.uniformity(Coverage)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TEQC)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: IRanges
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: GenomicRanges
Loading required package: Biostrings
Loading required package: XVector
Loading required package: hwriter
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TEQC/coverage.uniformity.Rd_%03d_medium.png", width=480, height=480)
> ### Name: coverage.uniformity
> ### Title: Coverage uniformity plot
> ### Aliases: coverage.uniformity
> ### Keywords: hplot
>
> ### ** Examples
>
> ## get reads and targets
> exptPath <- system.file("extdata", package="TEQC")
> readsfile <- file.path(exptPath, "ExampleSet_Reads.bed")
> reads <- get.reads(readsfile, idcol=4, skip=0)
[1] "read 19546 sequenced reads"
> targetsfile <- file.path(exptPath, "ExampleSet_Targets.bed")
> targets <- get.targets(targetsfile, skip=0)
[1] "read 50 (non-overlapping) target regions"
Warning message:
the "reduce" method for RangedData object is deprecated
>
> ## calculate per-base coverages
> Coverage <- coverage.target(reads, targets, perBase=TRUE)
>
> ## coverage uniformity plot
> coverage.uniformity(Coverage)
>
>
>
>
>
> dev.off()
null device
1
>
|