Import mutation profiles from a Manual Annotation Format (MAF) file. All mutations are aggregated as a
unique event type labeled "Mutation" and assigned a color according to the default of function
import.genotypes. If this is a TCGA MAF file check for multiple samples per patient is performed
and a warning is raised if these occurr. Customized MAF files can be imported as well provided that
they have columns Hugo_Symbol, Tumor_Sample_Barcode and Variant_Classification.
Custom filters are possible (via filter.fun) to avoid loading the full MAF data
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TRONCO)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TRONCO/import.MAF.Rd_%03d_medium.png", width=480, height=480)
> ### Name: import.MAF
> ### Title: import.MAF
> ### Aliases: import.MAF
>
> ### ** Examples
>
> data(maf)
> mutations = import.MAF(maf)
*** Importing from dataframe
Loading MAF dataframe ...DONE
*** Mutations names: using Hugo_Symbol
*** Using full MAF: #entries 16
*** MAF report: TCGA=TRUE
Type of annotated mutations:
[1] Missense_Mutation Silent Nonsense_Mutation Frame_Shift_Ins
Levels: Frame_Shift_Ins Missense_Mutation Nonsense_Mutation Silent
*** [merge.mutation.types = T] Mutations will be merged and annotated as 'Mutation'
Number of samples: 3
[TCGA = TRUE] Number of TCGA patients: 3
Number of annotated mutations: 16
Mutations annotated with "Valid" flag (%): 88
Number of genes (Hugo_Symbol): 13
Starting conversion from MAF to 0/1 mutation profiles (1 = mutation) :3 x 13
................
Starting conversion from MAF to TRONCO data type.
> mutations = annotate.description(mutations, 'Example MAF')
> mutations = TCGA.shorten.barcodes(mutations)
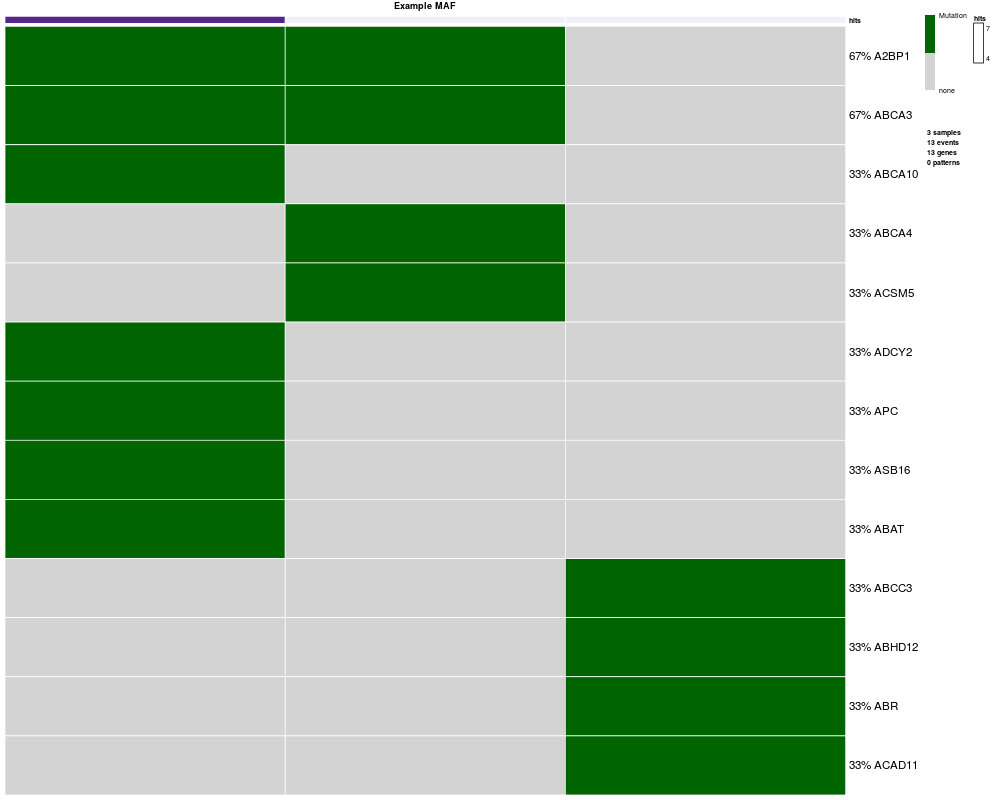
> oncoprint(mutations)
*** Oncoprint for "Example MAF"
with attributes: stage = FALSE, hits = TRUE
Sorting samples ordering to enhance exclusivity patterns.
Setting automatic row font (exponential scaling): 11.6
>
>
>
>
>
>
> dev.off()
null device
1
>
 .
.