Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Transcription Start Site IdentificationDescriptionThe TSSi package normalizes and identifies transcription start sites in high-throughput sequencing data. DetailsHigh throughput sequencing has become an essential experimental approach for the investigation of transcriptional mechanisms. For some applications like ChIP-seq, there are several available approaches for the prediction of peak locations. However, these methods are not designed for the identification of transcription start sites (TSS) because such data sets have qualitatively different noise. The TSSi provides a heuristic framework for the identification of TSS based on high-throughput sequencing data. Probabilistic assumptions for the count distribution as well as for systematic errors, i.e. for contaminating measurements close to a TSS, are made and can be adapted by the user. The framework also comprises a regularization procedure which can be applied as a preprocessing step to decrease the noise and thereby reduce the number of false predictions. The package is published under the GPL-3 license. Author(s)Clemens Kreutz, Julian Gehring, Jens Timmer Maintainer: Julian Gehring <julian.gehring@fdm.uni-freiburg.de> ReferencesC. Kreutz, J. Gehring, D. Lang, J. Timmer, and S. Rensing: TSSi - An R package for transcription start site identification from high throughput sequencing data. in preparation See AlsoPackage:
Methods:
Functions:
Classes:
Data set:
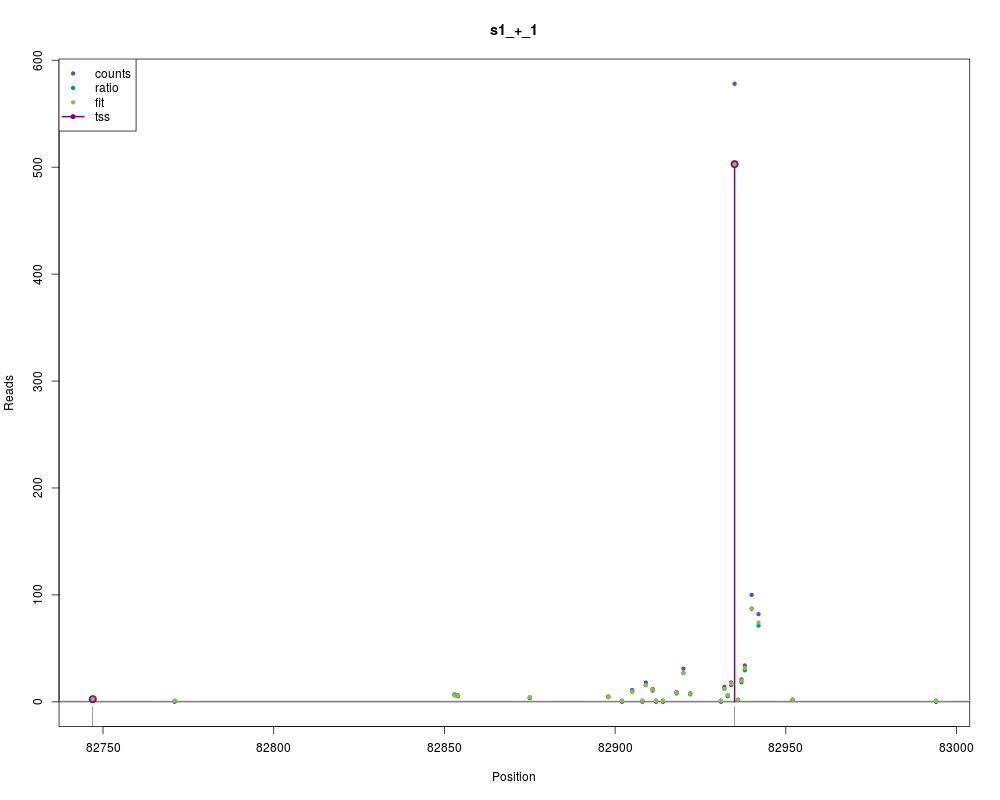
Examples## load data set data(physcoCounts) ## segmentize data attach(physcoCounts) x <- segmentizeCounts(counts=counts, start=start, chr=chromosome, region=region, strand=strand) detach(physcoCounts) x segments(x) ## normalize data, w/o and w/ fitting yRatio <- normalizeCounts(x) yFit <- normalizeCounts(x, fit=TRUE) yFit ## identify TSS z <- identifyStartSites(yFit) z ## inspect results head(tss(z, 1)) plot(z, 1) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TSSi)
Attaching package: 'TSSi'
The following object is masked from 'package:graphics':
segments
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TSSi/TSSi-package.Rd_%03d_medium.png", width=480, height=480)
> ### Name: TSSi-package
> ### Title: Transcription Start Site Identification
> ### Aliases: TSSi TSSi-package
> ### Keywords: package htest models
>
> ### ** Examples
>
> ## load data set
> data(physcoCounts)
>
> ## segmentize data
> attach(physcoCounts)
> x <- segmentizeCounts(counts=counts, start=start, chr=chromosome,
+ region=region, strand=strand)
> detach(physcoCounts)
>
> x
* Object of class 'TssData' *
Data imported
** Segments **
Segments (5): s1_+_1, s1_+_2, s1_-_3, s2_+_4, s2_-_5
Chromosomes (2): s1, s2
Strands (2): +, -
Regions (5): 1, 2, 3, 4, 5
nCounts (5): 978, 587, 848, 466, 690
> segments(x)
chr strand region nPos nCounts start end
s1_+_1 s1 + 1 28 978 82747 82994
s1_+_2 s1 + 2 33 587 814741 815042
s1_-_3 s1 - 3 47 848 1435037 1435157
s2_+_4 s2 + 4 68 466 1454505 1455353
s2_-_5 s2 - 5 43 690 1574882 1575467
>
> ## normalize data, w/o and w/ fitting
> yRatio <- normalizeCounts(x)
> yFit <- normalizeCounts(x, fit=TRUE)
> yFit
* Object of class 'TssNorm' *
Data normalized
** Segments **
Segments (5): s1_+_1, s1_+_2, s1_-_3, s2_+_4, s2_-_5
Chromosomes (2): s1, s2
Strands (2): +, -
Regions (5): 1, 2, 3, 4, 5
nCounts (5): 978, 587, 848, 466, 690
** Parameters **
pattern: %1$s_%2$s_%3$s
offset: 10
basal: 1e-04
lambda: c(0.1, 0.1)
fit: TRUE
optimizer: all
>
> ## identify TSS
> z <- identifyStartSites(yFit)
> z
* Object of class 'TssResult' *
TSS in data identified
** Segments **
Segments (5): s1_+_1, s1_+_2, s1_-_3, s2_+_4, s2_-_5
Chromosomes (2): s1, s2
Strands (2): +, -
Regions (5): 1, 2, 3, 4, 5
nCounts (5): 978, 587, 848, 466, 690
nTSS (5): 2, 3, 1, 9, 6
** Parameters **
pattern: %1$s_%2$s_%3$s
offset: 10
basal: 1e-04
lambda: c(0.1, 0.1)
fit: TRUE
optimizer: all
tau: c(20, 20)
threshold: 1
fun: function (fg, bg, indTss, pos, basal, tau, extend = FALSE)
{
idx <- pos - pos[1] + 1
idxTss <- idx[indTss]
n <- pos[length(pos)] - pos[1] + 1
fak <- 1/.exppdf(1, tau)
win1 <- fak[1] * .exppdf(n:1, tau[1])
win2 <- fak[2] * .exppdf(1:n, tau[2])
win <- c(win1, 0, win2)
bgb <- rep(0, n)
bgb[idxTss] <- bg[indTss]
cums <- convolve(win, rev(bgb), type = "open")
expect <- cums[(n + 1):(length(cums) - n)]
if (!extend)
expect <- expect[idx]
expect[expect < basal] <- basal
delta <- fg - expect
delta[delta < 0] <- 0
res <- list(delta = delta, expect = expect)
return(res)
}
readCol: fit
neighbor: TRUE
>
> ## inspect results
> head(tss(z, 1))
pos reads
1 82747 2.551251
2 82935 502.836991
> plot(z, 1)
>
>
>
>
>
> dev.off()
null device
1
>
|