Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Plot read profiles for a particular genomic region.Description
Usage
plotRegion(object, region, seqname, SNPs = TRUE, xlab = "", title = "",
size = 0.5, BPPARAM = bpparam())
## S4 method for signature 'TargetExperiment'
plotRegion(object, region, seqname, SNPs = TRUE,
xlab = "", title = "", size = 0.5, BPPARAM = bpparam())
Arguments
Valueggplot2 graphics. include TargetExperiment-FeatPerform.R Notesee full example in Author(s)Gabriela A. Merino gmerino@bdmg.com.ar, Cristobal Fresno cfresno@bdmg.com.ar and Elmer A. Fernandez efernandez@bdmg.com.ar See Also
Examples
if(interactive()){
## loading TargetExperiment object
data(ampliPanel, package="TarSeqQC")
## Defining bam file, bed file and fasta file names and paths
setBamFile(ampliPanel)<-system.file("extdata", "mybam.bam",
package="TarSeqQC", mustWork=TRUE)
setFastaFile(ampliPanel)<-system.file("extdata", "myfasta.fa",
package="TarSeqQC", mustWork=TRUE)
# getting and exploring a sequenced region of a particular gene
getRegion(ampliPanel, level="gene", ID="gene7", collapse=FALSE)
# plot a particular genomic region
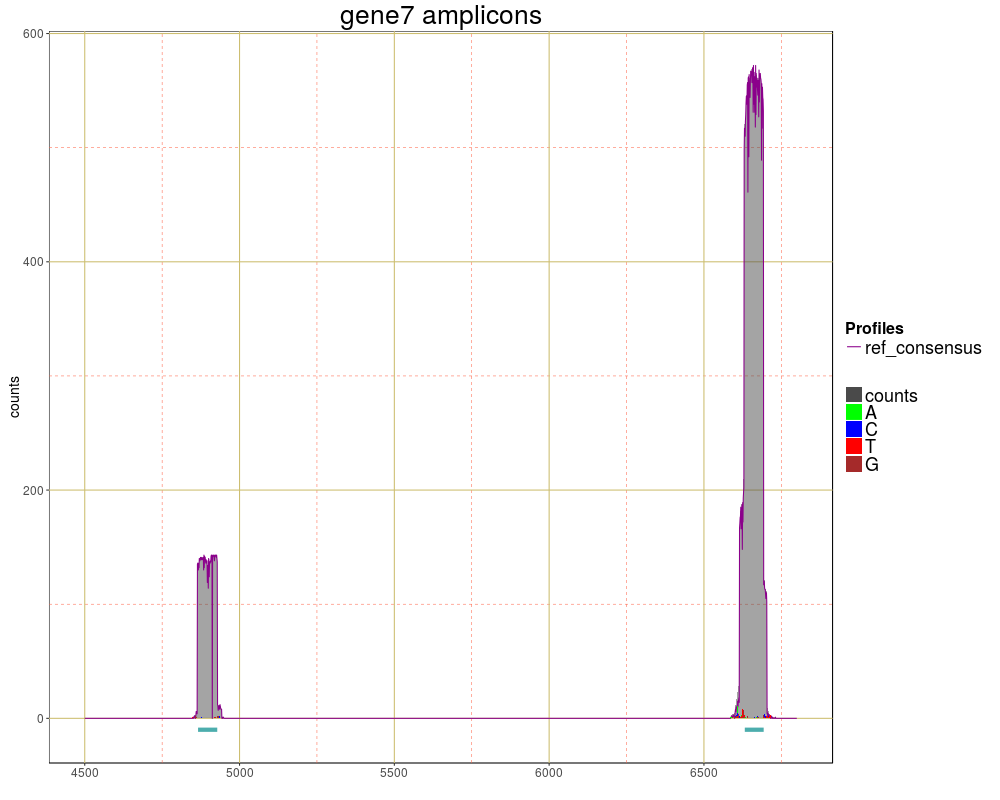
g<-plotRegion(ampliPanel,region=c(4500,6800), seqname="chr10", SNPs=TRUE,
xlab="", title="gene7 amplicons",size=0.5)
# x11(type="cairo")
g
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(TarSeqQC)
Loading required package: GenomicRanges
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomeInfoDb
Loading required package: Rsamtools
Loading required package: Biostrings
Loading required package: XVector
Loading required package: ggplot2
Loading required package: plyr
Attaching package: 'plyr'
The following object is masked from 'package:XVector':
compact
The following object is masked from 'package:IRanges':
desc
The following object is masked from 'package:S4Vectors':
rename
Loading required package: openxlsx
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/TarSeqQC/TargetExperiment-plotRegion.Rd_%03d_medium.png", width=480, height=480)
> ### Name: plotRegion
> ### Title: Plot read profiles for a particular genomic region.
> ### Aliases: plotRegion plotRegion,TargetExperiment-method
> ### plotRegion-methods
>
> ### ** Examples
>
> #if(interactive()){
> ## loading TargetExperiment object
> data(ampliPanel, package="TarSeqQC")
> ## Defining bam file, bed file and fasta file names and paths
> setBamFile(ampliPanel)<-system.file("extdata", "mybam.bam",
+ package="TarSeqQC", mustWork=TRUE)
> setFastaFile(ampliPanel)<-system.file("extdata", "myfasta.fa",
+ package="TarSeqQC", mustWork=TRUE)
>
> # getting and exploring a sequenced region of a particular gene
> getRegion(ampliPanel, level="gene", ID="gene7", collapse=FALSE)
names seqname start end gene
1 AMPL18 chr10 141 233 gene7
2 AMPL19 chr10 1007 1079 gene7
3 AMPL20 chr10 4866 4928 gene7
4 AMPL21 chr10 6632 6693 gene7
5 AMPL22 chr10 8475 8527 gene7
> # plot a particular genomic region
> g<-plotRegion(ampliPanel,region=c(4500,6800), seqname="chr10", SNPs=TRUE,
+ xlab="", title="gene7 amplicons",size=0.5)
> # x11(type="cairo")
> g
> #}
>
>
>
>
>
> dev.off()
null device
1
>
|