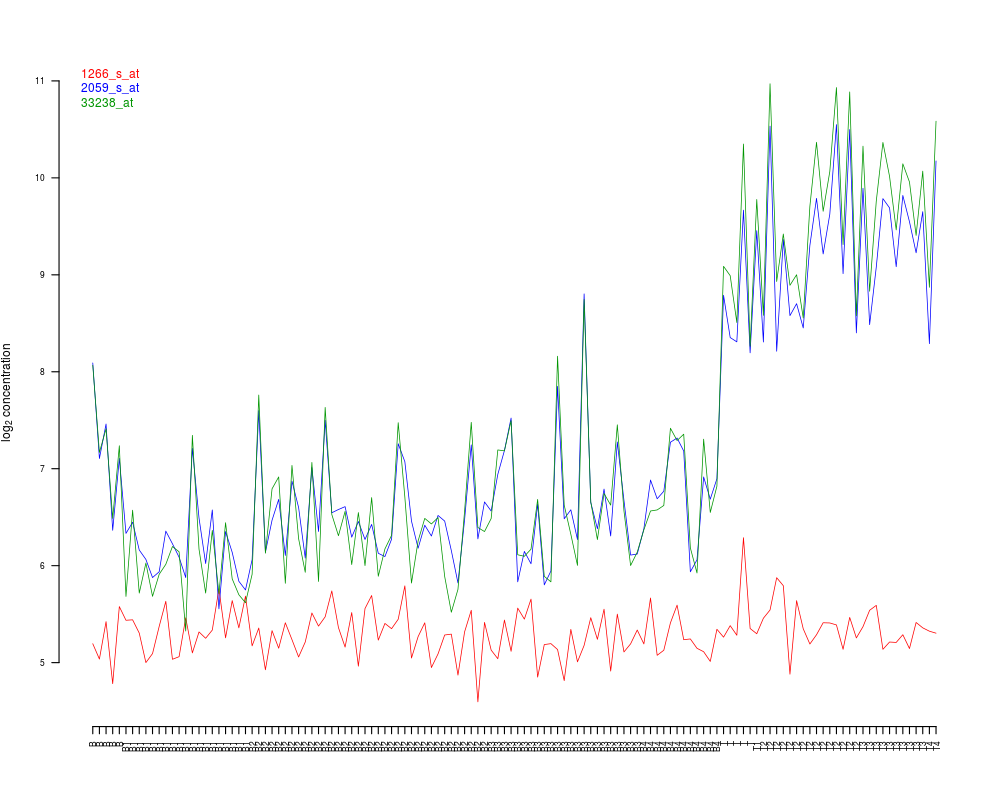
The probeset ID. These should be stored in the featureNames
of the expressionSet object.
colvec
Vector of colors to be used for the groups. If not specified, the default colors of
a4palette are used.
sampleIDs
A boolean or a string to determine the labels on the x-axis. Setting it to FALSE
results in no labels (interesting when the labels are unreadable due to large sample sizes).
Setting it to a string will put the values of that particular pData column as labels.
The string should be a name of a column in the pData of the expressionSet object."
addLegend
Boolean indicating whether a legend for the colors of the dots should be added.
legendPos
Specify where the legend should be placed. Typically either topright,
bottomright, topleft (the default) or bottomleft
orderGroups
String containing the name of the grouping variable to order the samples
in the x-axis accordingly. This should be a name of a column in the pData of
the expressionSet object.
...
Possibility to add extra plot options. See par
Author(s)
W. Talloen
See Also
plot1gene, boxPlot
Examples
if (require(ALL)){
data(ALL, package = "ALL")
ALL <- addGeneInfo(ALL)
ALL$BTtype <- as.factor(substr(ALL$BT,0,1))
myGeneSymbol <- c("LCK") # a gene
probesetPos <- which(myGeneSymbol == featureData(ALL)$SYMBOL)
myProbesetIds <- featureNames(ALL)[probesetPos]
profilesPlot(object = ALL, probesetIds = myProbesetIds,
orderGroups = "BT", sampleIDs = "BT")
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(a4Base)
Loading required package: grid
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: AnnotationDbi
Loading required package: stats4
Loading required package: IRanges
Loading required package: S4Vectors
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: annaffy
Loading required package: GO.db
Loading required package: KEGG.db
KEGG.db contains mappings based on older data because the original
resource was removed from the the public domain before the most
recent update was produced. This package should now be considered
deprecated and future versions of Bioconductor may not have it
available. Users who want more current data are encouraged to look
at the KEGGREST or reactome.db packages
Loading required package: mpm
Loading required package: MASS
Attaching package: 'MASS'
The following object is masked from 'package:AnnotationDbi':
select
Loading required package: KernSmooth
KernSmooth 2.23 loaded
Copyright M. P. Wand 1997-2009
mpm version 1.0-22
Loading required package: genefilter
Attaching package: 'genefilter'
The following object is masked from 'package:MASS':
area
Loading required package: limma
Attaching package: 'limma'
The following object is masked from 'package:BiocGenerics':
plotMA
Loading required package: multtest
Loading required package: glmnet
Loading required package: Matrix
Attaching package: 'Matrix'
The following object is masked from 'package:S4Vectors':
expand
Loading required package: foreach
Loaded glmnet 2.0-5
Loading required package: a4Preproc
Loading required package: a4Core
Attaching package: 'a4Core'
The following object is masked from 'package:limma':
topTable
Loading required package: gplots
Attaching package: 'gplots'
The following object is masked from 'package:multtest':
wapply
The following object is masked from 'package:IRanges':
space
The following object is masked from 'package:S4Vectors':
space
The following object is masked from 'package:stats':
lowess
a4Base version 1.20.0
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/a4Base/profilesPlot.Rd_%03d_medium.png", width=480, height=480)
> ### Name: profilesPlot
> ### Title: Plot expression profiles of multiple genes or probesets
> ### Aliases: profilesPlot
>
> ### ** Examples
>
> if (require(ALL)){
+ data(ALL, package = "ALL")
+ ALL <- addGeneInfo(ALL)
+ ALL$BTtype <- as.factor(substr(ALL$BT,0,1))
+
+ myGeneSymbol <- c("LCK") # a gene
+ probesetPos <- which(myGeneSymbol == featureData(ALL)$SYMBOL)
+ myProbesetIds <- featureNames(ALL)[probesetPos]
+
+ profilesPlot(object = ALL, probesetIds = myProbesetIds,
+ orderGroups = "BT", sampleIDs = "BT")
+ }
Loading required package: ALL
Loading required package: hgu95av2.db
Loading required package: org.Hs.eg.db
>
>
>
>
>
> dev.off()
null device
1
>
 .
.